
SRY-negative 45,X/46,XY adult male with complete masculinization and infertility: A case report and review of literature
2020-04-07 02:13:18YanHuaWuKeNaSunHuiBaoYingJianChen
World Journal of Clinical Cases 2020年24期
Yan-Hua Wu, Ke-Na Sun, Hui Bao, Ying-Jian Chen
Yan-Hua Wu, Hui Bao, Ying-Jian Chen, Department of Laboratory Medicine, The 960th Hospital of The PLA Joint Logistics Support Force, Jinan 250031, Shandong Province, China
Ke-Na Sun, Department of Medical Laboratory, Weifang Medical University, Weifang 261053,Shandong Province, China
Abstract BACKGROUND 45,X/46,XY mosaicism is a rare chromosomal abnormality with a wide range of phenotypes in both males and females, from normal individuals with different degrees of genital ambiguity to those who show signs of Turner’s syndrome.More rarely, cases of 45,X/46,XY mosaicism with a normal-appearing male phenotype are not found until a chromosome test is performed to investigate the cause of male infertility.CASE SUMMARY In this study, a 29-year-old male patient with complete azoospermia is reported.Chromosomal analyses of his lymphocytes revealed the karyotype 45,X[93%]/46,X,+mar(Y)[7%]. In addition, Y chromosome-specific markers, such as SRY,ZFY, AZFa, AZFb and AZFc, were not observed in his blood DNA according to multiplex polymerase chain reaction test. A literature review identified several 45,X/46,XY cases with a normal-appearing male phenotype, most of whom were diagnosed during infertility investigation. However, the present case is the first SRY-negative 45,X/46,XY male case diagnosed during a premarital medical examination.CONCLUSION This finding further suggests that sex determination is a complex process regulated by multiple genetic and environmental factors.
Key Words: Azoospermia; Sex chromosome; Mosaicism; Y chromosomal microdeletions;SRY-negative; Case report
INTRODUCTION
As a rare complement, the chromosomal abnormality of 45,X/46,XY mosaicism is found in 1.7 cases among 10000 newborns[1]. The spectrum of observed phenotypes ranges continuously from normal individuals with varying degrees of genital ambiguity to Turner’s syndrome[2]. Most normal-appearing male phenotype cases are diagnosed during the prenatal period, and cases with genital/gonadal anomalies are usually diagnosed after birth[3]. More rarely, cases of 45,X/46,XY mosaicism with a normal-appearing male phenotype are not found until a chromosome test is performed to investigate the cause of male infertility.
Because the Y chromosome carries testis-determining factor, which is a genetically predominant locus, under normal circumstances, the bipotent gonadal primordium can be triggered and testes formation can be processed, which makes the Y chromosome a key factor in human sex determination. TheSRYgene located in Yp11.2 was found to be cytogenetic and confirmed to play a critical role in the complex and tightly regulated processes of testis development[4]and sex differentiation[5]. However,more and moreSRY-negative male cases with various karyotypes have been reported.In addition, increasing studies have shown that other factors, including both genetic and environmental factors, may regulate gender determination and differentiation through a multi-target approach.
Key genetic factors are known to regulate spermatogenesis on Yq, namely azoospermia factors (AZFs), includingAZFa,AZFb andAZFc. Spermatogenetic failure caused byAZFmicrodeletions is a common cause of male infertility. Studies have shown thatAZFmicrodeletions can be detected in approximately 10%-15% of azoospermia patients in China[6].
In the present report, we describe the diagnosis of a rareSRY-negative male case with 45,X/46,XY mosaicism. In addition, we review the 45,X/46,XY male phenotype cases reported in the literature to date to provide a more comprehensive description of the genetic and pathological features of this subgroup.
CASE PRESENTATION
Chief complaints
A 29-year-old man visited our urology clinic for a premarital medical examination,with complaints of occasional scrotal pain.
History of present illness
For the previous month, the patient had experienced occasional minor pain in the testicles.
History of past illness
The patient had no notable previous medical history.
Personal and family history
He denied any family history and had no specific past history.
Physical examination
His height was 167 cm, and his weight was 57.9 kg. After physical examination, we found that he had no dysmorphisms and had a normal distribution of pubic hair and body hair. His external urethral meatus was in a normal position, and his penis had a normal appearance and size (5.7 cm, non-erectile).
Laboratory examinations
The results of the patient's serum test revealed that the luteinizing hormone (LH)concentration was elevated at 15.73 IU/L (normal range: 1.7-8.6 IU/L), and the folliclestimulating hormone (FSH) concentration was elevated at 14.13 IU/L (normal range:1.5-12.4 IU/L). However, the serum testosterone hormone concentration was 3.22 μg/L, which was in the normal range for adult males of 2.49-8.36 μg/L. Azoospermia was determined after repeated seminal analysis. Chromosomal analysis was performed twice on samples collected at different times, and 100 metaphases were analyzed in each analysis. Two different cell lines with the karyotype 45,X[93%]/46,X,+mar(Y)[7%] were observed by GTG banding. Fluorescencein situhybridization analysis with screening of metaphase and interphase lymphocytes was carried out to confirm the result of the karyotype analysis. Two cell lines, one with one green signal for Xcen (182/200) and the other with one green signal and one red signal for Xcen and Ycen (18/200), respectively, were observed according to fluorescencein situhybridization (Figure 1A and B). All the metaphase and interphase lymphocytes showed one signal for Xcen but noSRYsignal, except for cell lines containingSRY(Figure 1C). Polymerase chain reaction amplification of 16Y-STSgene loci (SRY,ZFY,sY86,sY84,CDY2,SMCY,sY127,sY134,sY1161,sY1191,sY254,sY255,DAZ,sY157,CDY1,ZFX,SMCX,DAZL) using a Y-chromosome microdeletion detection kit(Microread Gene; Beijing, China) demonstrated the presence of Y chromosomederived sequences. TheSRYandZFYgenes were not amplified in theAZFregion(Figure 1D). The negative amplification ofSRYfurther confirmed the partial absence of the Y-chromosome sequence.
Imaging examinations
Ultrasound scanning of the scrotum showed that both testicles were located in the scrotum, but the volumes (6.6 mL and 6.8 mL, respectively) were significantly smaller than the normal adult male testicle size (range: 15-23 mL). In addition, a normal-sized prostate and seminal vesicles were observed by internal genitalia ultrasound analysis.
FINAL DIAGNOSIS
Azoospermia.
TREATMENT
The recommended treatments were hormone replacement therapy, including oral testosterone undecanoate to maintain sexual function, and sperm donation and assisted reproductive technology to solve fertility problems.
OUTCOME AND FOLLOW-UP
Follow-up found that the patient had a normal penile erection. He was married 1 year later, and the couple decided to adopt a child after marriage.
DISCUSSION
Stability of the number and structure of chromosomes is the basic requirement for maintaining the normal sex differentiation process. Two decades ago, Telviet al[3]found that 45,X/46,XY mosaicism can manifest as a normal male phenotype and can also cause some abnormal clinical phenotypes, including Turner’s syndrome,pseudohermaphroditism, and mixed gonad dysplasia. The subgroup of 45,X/46,XY mosaicism with normal adult male phenotype is usually diagnosed during infertility investigation. Lashkariet al[7]reported that the occurrence rate of 45,X/46,XY mosaicism in azoospermic and oligozoospermic patients was 0.78%. Among the 49 infertile adult male patients with the 45,X/46,XY mosaicism karyotype that they evaluated, 21 showed azoospermia, 24 had sperm abnormalities, and four displayed a normal spermogram.
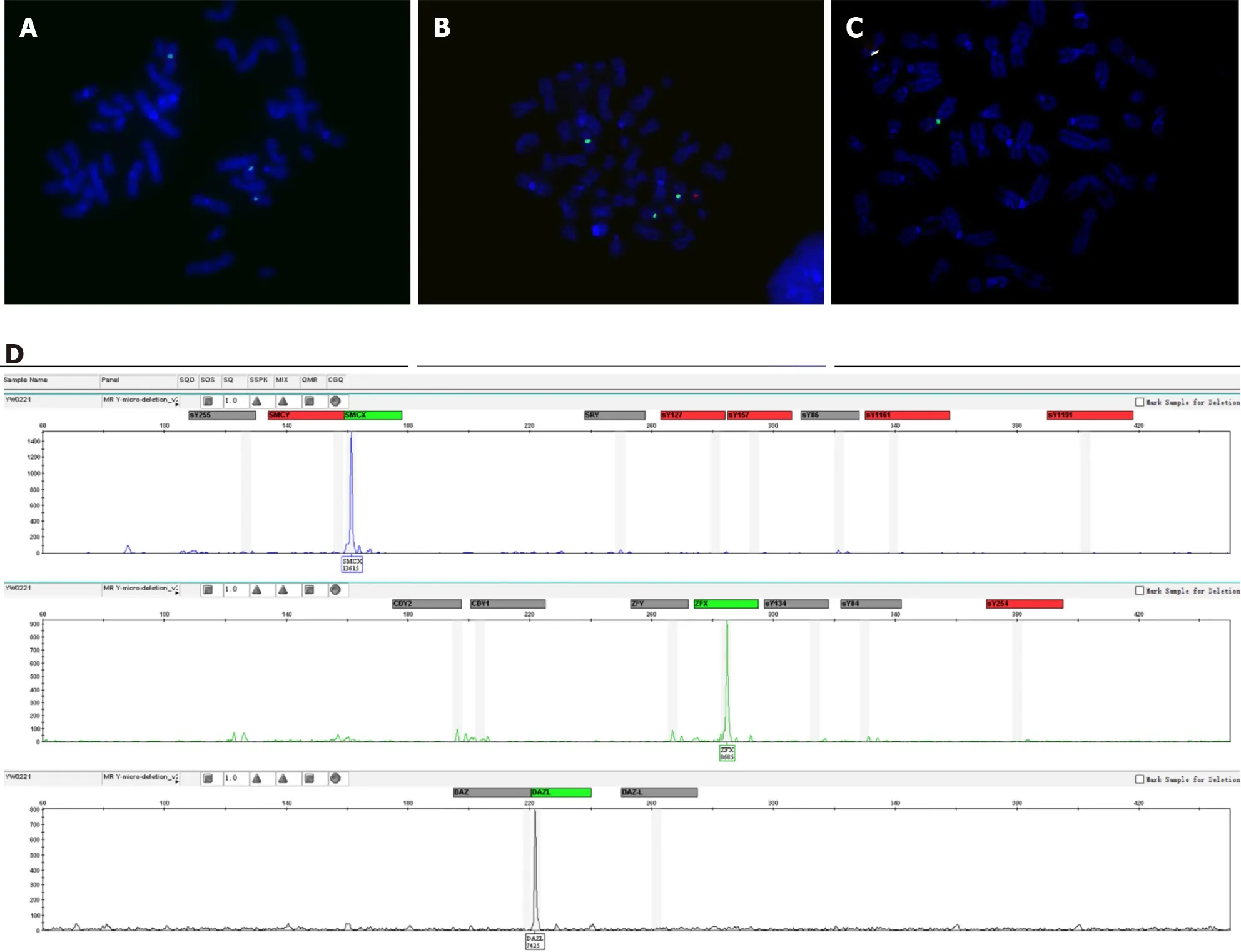
Figure 1 Fluorescence in situ hybridization analysis of metaphase chromosomes. A-C: Fluorescence in situ hybridization analysis showed the presence of two cell lines, one with one green signal for Xcen (A) and the other with one green signal and one red signal for Xcen and Ycen, respectively (B), as well as the absence of the SRY gene, with only green signal of the X chromosome (C); D: Polymerase chain reaction amplification of 16 Y-STS gene loci.
We reviewed additional literature regarding 45,X/46,XY adult male cases(Table 1)[8-18]. Among the 34 cases reviewed, 96.5% (28/29) showed azoospermia or oligozoospermia. The 45,X/46,XY mosaicism rates ranged from 6/93.3 to 94/6.7. There was no relationship between the mosaicism rate in peripheral blood lymphocyte and the phenotype, which was consistent with previous results[19,20]. The mosaicism ratio in different tissues may explain the variety of phenotypes in mixed gonadal dysgenesis[21]. Moreover, these individuals showed short or normal stature, with height ranged from 148-181 cm (data from 24 cases). Most of the patients had small testicles (92%, 23/25), elevated LH concentration (65.2%, 15/23), elevated FSH concentration (80.9%, 17/21), and normal testosterone concentration (95.2%, 20/21).All patients had theSRYgene, while 62.1% (18/29) had theAZFmicrodeletion. The most commonAZFmicrodeletions wereAZF(b+c) (66.7%, 12/18) followed byAZFc(22.2%, 4/12),AZFb (5.5%, 1/18), andAZF(a+b+c) (5.5%, 1/18). In our study, the man with the 45,X/46,XY mosaic karyotype also showed complete masculinization and azoospermia, with short stature, small testicles, elevated LH and FSH concentrations,and a normal T concentration. However, theSRYgene,ZFYgene, andAZF(a+b+c)regions of blood DNA were missing in this case, and this is the first male with theSRY-negative 45,X/46,XY mosaic karyotype according to our literature search results.
The process of sexual differentiation begins in the early stages of human embryo development. After a series of complex and orderly procedures, bipotential gonads eventually develop into testes or ovaries. TheSRYgene plays a critical role in the cascade of events of sexual differentiation. The histogenesis of testis is initiated by theSRYgene, beginning at about 6 wk post-implantation[21,22]. On the other hand, a deletion mutant of theSRYgene can affect masculinization and may cause 46,XY female sex reversal[23]. Even though theSRYgene is critical in the initiation of testis determination, some of theSRY-negative phenotype may show the typical male phenotype, as seen in the present patient. Our findings further suggest that testicular formation and development occursviaa comprehensive process jointly regulated by other key genetic factors or environmental factors in addition to theSRYgene. Several hypotheses have attempted to explain rationally the formation of testicles inSRYnegative males, such as the possible predominance of the 46,XY cell line in the gonads[20], hidden mosaicism for a Y-derivative material, or mutation of an autosomal or X-chromosomal gene downstream fromSRY. In addition, studies have shown that overexpression of theSOX9gene can initiate testis differentiation when theSRYgene is silenced. This result indicates that theSOX9gene, as downstream factor ofSRY,plays an important role in the sex determination process[24-26]. In the present study, the patient refused to undergo genetic analysis of genital skin and gonadal fibroblasts as well as further examinations, and thus, the exact mechanism of his gender development could not be explained.
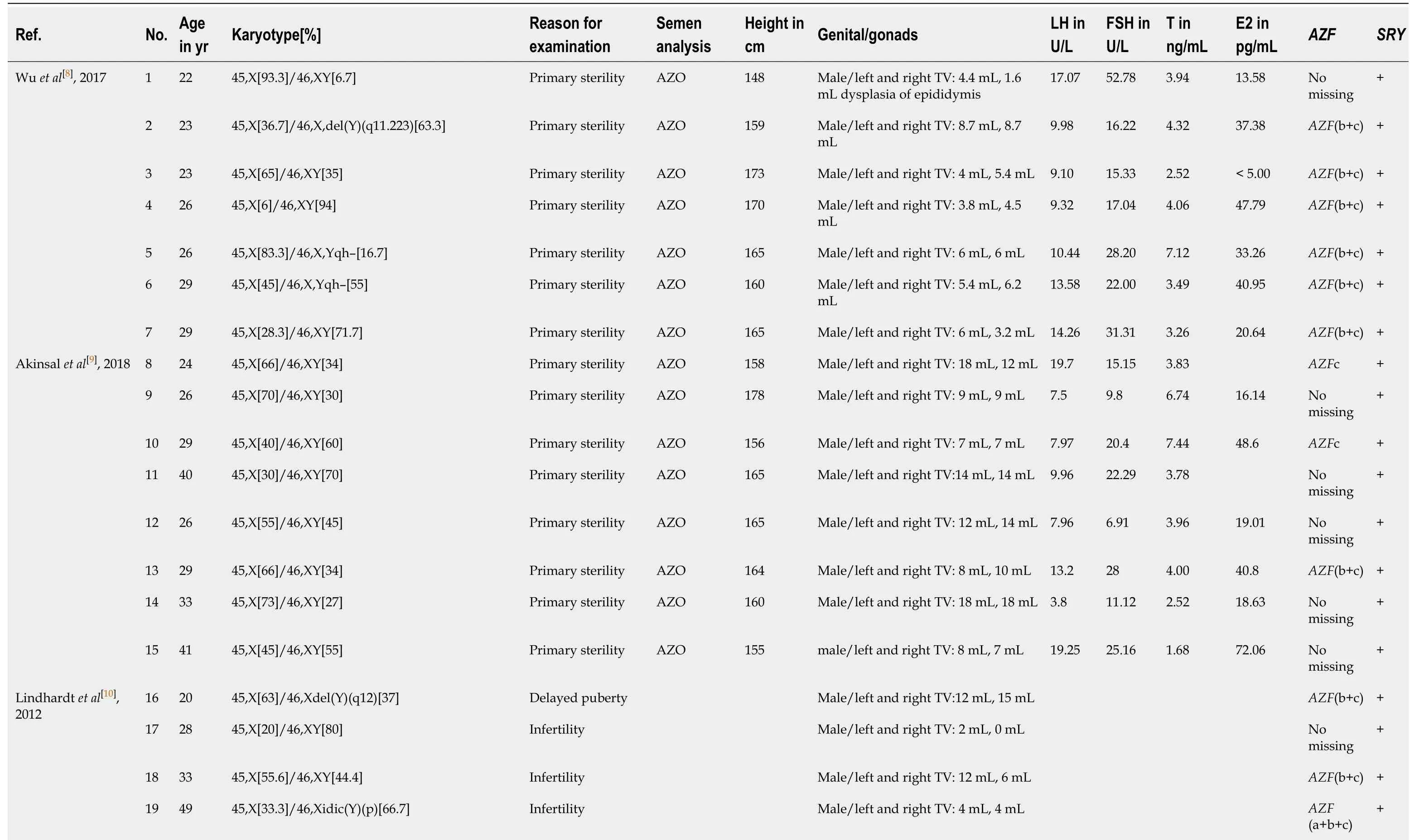
Table 1 Characteristics of 34 reported 45,X/46,XY adult male cases
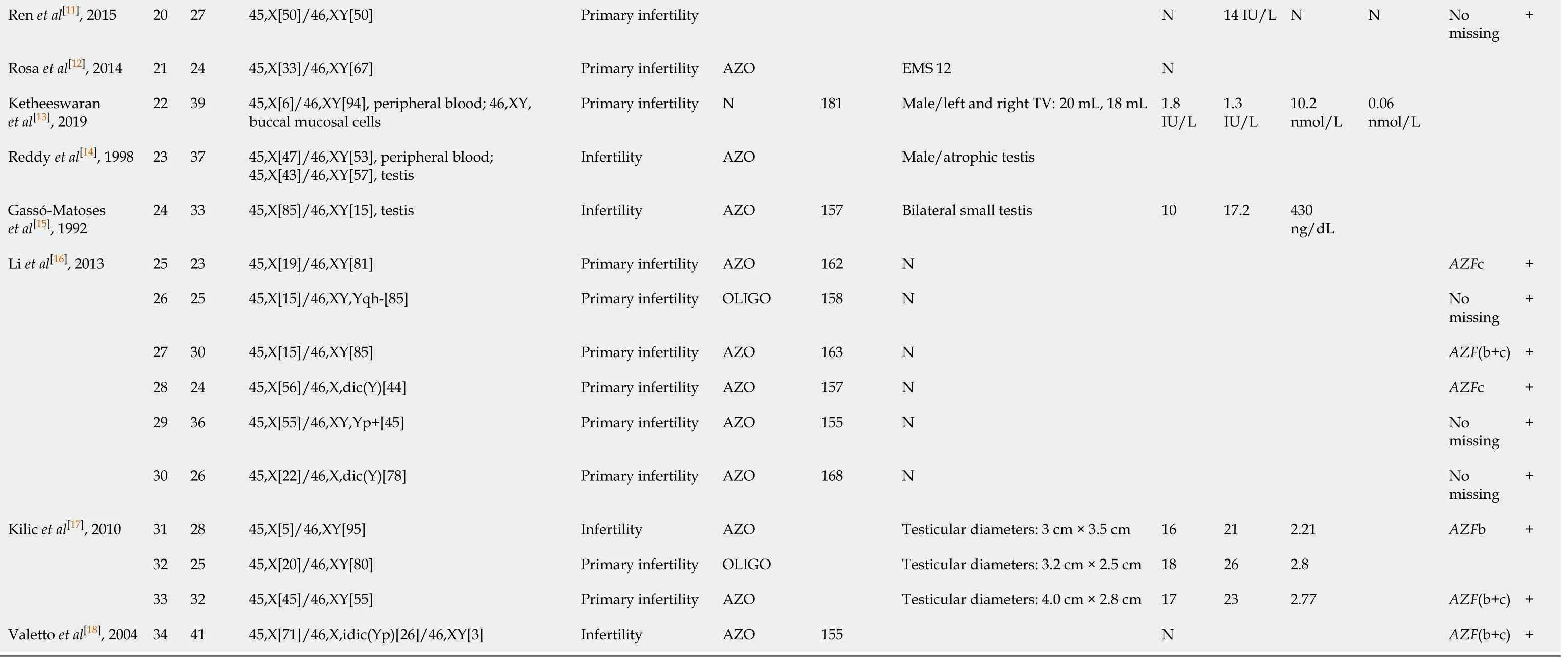
AZO: Azoospermia; E2: Estrogen; EMS: External masculinization score; FSH: Follicle-stimulating hormone; LH: Luteinizing hormone; N: Normal; OLIGO: Oligozoospermia; T: Testosterone; TV: Testicular volume.
TheAZFgene is located on the long arm of the Y chromosome, and its locus contains protein-coding genes essential for spermatogenesis[27]. Y chromosome microdeletion, which might result from Y-chromosome instability and lead to 45,X karyotype, is one of the key causes of severe male infertility. Clinical statistics indicate that 10%-15% of azoospermic patients and 5%-10% of severe oligospermia patients have Y chromosome microdeletion[6,28]. Studies have reported that deletion of large and submicroscopic Y chromosome may lead to an increased proportion of 45,X abnormal karyotype cells among sperm cells and lymphocytes[29]. In the present case, the patient had deletions ofAZF(a+b+c) regions in addition to 45,X/46, XY mosaicism, which may explain the high percentage of 45,X cells and azoospermia.
From the perspective of oncology, gonadal germ cell tumors are detected at an elevated frequency among patients with a 45,X/46,XY karyotype and malformations of the external genitalia[30]. The risk for malignant transformation is reportedly about 10% and increases with age in patients with 45,X/46,XY gonadal dysgenesis[31,32]. It is worth noting that the prognoses of 45,X/46,XY patients with an apparently normal male phenotype until adulthood and patients who are born with severe genital anomalies show no statistical difference[33]. Therefore, adult patients with the 45,X/46,XY mosaic karyotype must be followed up for life, with particular focus on testicular function and testicular tumor screening.
CONCLUSION
In conclusion, we have described the clinical and genetic findings for a male with complete virilization inSRY-negative 45,X/46,XY mosaicism. We believe that a perfect karyotype analysis and Y-microdeletion analysis could not only reveal the cause of male infertility, in order to facilitate reproductive counseling, but also provide prognostic information for patients with specific karyotypes.
 World Journal of Clinical Cases2020年24期
World Journal of Clinical Cases2020年24期
- World Journal of Clinical Cases的其它文章
- Primary duodenal tuberculosis misdiagnosed as tumor by imaging examination: A case report
- Successful endovascular treatment with long-term antibiotic therapy for infectious pseudoaneurysm due to Klebsiella pneumoniae: A case report
- Idiopathic adulthood ductopenia with elevated transaminase only: A case report
- Takotsubo cardiomyopathy associated with bronchoscopic operation: A case report
- Extracorporeal shock wave therapy treatment of painful hematoma in the calf: A case report
- Rare case of drain-site hernia after laparoscopic surgery and a novel strategy of prevention: A case report