
液相色谱-质谱/质谱法测定大米中莠去津残留量的不确定度评定
2020-03-06 16:34:30蒋国振
粮食与食品工业 2020年1期
刘 萍,刘 旭,王 婧,蒋国振
陕西省粮油产品质量监督检验中心 (西安 710016)
我国人口中有65%以大米为主食,大米在国家粮食安全中的地位举足轻重。莠去津(atrazine)是内吸选择性苗前、苗后封闭除草剂,主要作用于双子叶植物,对大草效果比较不理想,是大米常用农药。我国现行国家标准GB2763—2019《食品安全国家标准 食品中农药最大残留限量》中规定,谷物中莠去津限量指标为0.05 mg/kg。
本文根据GB/T20770—2008《粮谷中486种农药及相关化学品残留量的测定 液相色谱-串联质谱法》、《化学分析中的不确定度的评估指南》(CNAS—GL06)[1]、国家计量技术规范《测量不确定度评定与表示》[2],对大米中莠去津残留量测定的各个分量和最终结果的进行评定,以保证莠去津残留量测定方法的可靠性及测定结果的可信度。
1 材料与方法
1.1 仪器试剂
Waters Aquity I-Class/TQD 液相色谱-质谱/质谱仪;超声发生器(昆山,KQ2200DE 型);离心机(HERMLE,Z323);涡旋机(EYELA,CM-1000);电子天平(梅特勒,AB204-S);莠去津标准品(农业部环境保护科研检测所,丙酮配制,100μg/mL)。
1.2 方法
1.2.1 仪器条件
1.2.1.1 色谱条件
色谱柱,Waters Aquity UPLC BEH C18色谱柱(50 mm×2.1 mm,1.7μm);柱温25 ℃;进样量2μL;流速0.3 mL/min;流动相A(0.1%甲酸水溶液),B(乙腈),流动相梯度洗脱条件见表1。

时间/min流速/(mL/min)A0.1%甲酸水溶液/%B 乙腈/%0 0.3 99 1.0 1 0.3 70 30 2 0.3 60 40 4.5 0.3 40 60 6 0.3 1.0 99 7 0.3 99 1.0
1.2.1.2 质谱条件
离子源:电喷雾正离子(ESI+)电离模式;电喷雾电压3.5 kV;去溶剂气温度500 ℃;雾化气流量800 L/h;监测模式:多重反应监测(MRM)模式,定量离子对216.0/174.0,锥孔电压34 V,碰撞能量19 eV。
1.2.2 实验步骤
称取粉碎混匀的大米样品5 g(精确至0.01 g)于50 mL 离心管中,加入2.0g 无水硫酸钠和10 mL乙腈提取液,在振荡器上振荡提取10 min,再超声提取10 min,5000 rpm 离心5 min。取2.0mL上层提取液,经C18 固相萃取小柱净化,滤液经0.22μm滤膜过滤于样品瓶中,供HPLC-MS/MS测定。
1.2.3 标准曲线制备
准确吸取1.0mL莠去津标准溶液到10 mL容量瓶中,用甲醇溶液稀释并定容,得浓度为10μg/mL的莠去津储备溶液,储存于-20℃冰箱中。
配制莠去津标准曲线浓度为:2.0、20、50、100、200 ng/mL。进样2μL,以标准溶液浓度与检测离子峰面积作标准曲线。标准曲线Y=657.172×X+227.845,相关系数大于0.999。
2 结果与讨论
2.1 数学模型
测量不确定度数学模型如下所示:

式中:X 为试样中被测组分残留量,μg/kg;C为样品溶液中被测组分的浓度,ng/mL;V 为样品溶液最终定容体积,mL;m 为样品溶液所代表最终试样的质量,g。
2.2 不确定度来源分析
根据测量过程和数学模型分析,测定莠去津的不确定度主要有以下几方面:样品制备引入的不确定度分量;标准溶液配制引入的不确定度分量;仪器引入的不确定度分量;回收实验引入的不确定度分量。
2.3 制备样品溶液的相对不确定度(B类评定)
2.3.1 试样称量引入的相对标准不确定度ur(m)
称样量为5 g,实验所用天平的检定证书给出了最大允许误差为±0.05 g,取矩形分布[1],则试样称量引入的标准不确定度为:

称量试样的值为5.02 g,故试样称量引入的相对标准不确定度为:

2.3.2 样品前处理中提取、净化过程中体积引入的相对标准不确定度
(1)移液管引入的标准不确定度
10mL移液管引入的不确定度,按照JJG196-2006《常用玻璃量器检定规程》[3],10mL A 级移液管最大容量允差为±0.015 mL,取三角分布,则样品定容引入的标准不确定度u(V)为:

(2)温度波动引入的标准不确定度
实验过程中实验室温度控制在(20±4)℃,假定温度分布为矩形分布,乙腈的膨胀系数为1.37×10-3/℃,温度影响不确定度为:

相对标准不确定度:

配制样品溶液体积的相对不确定度为:

故样品制备引入的相对不确定度:

2.4 标准溶液制备引入的相对不确定度(B类评定)
2.4.1 标准物质引入的不确定度
本实验采用的莠去津标样购自农业部环境保护科研检测所,标准值为100μg/mL,扩展不确定度0.14μg/mL,引入的不确定度假定为矩形分布。相对不确定度:

2.4.2 标准溶液配制过程中量器自身引入的不确定度
根据JJG 196—2006 《常用玻璃量器检定规程》[3]、JJG646—2006《移液器》[4]要求,按照矩形分布处理,标准溶液配制过程中用到的玻璃量器和移液器引入的相对标准不确定度如表2所示。

影响因素 项目移液器 容量瓶5 mL 1 mL 200μL 20μL 10mL量取体积/mL 5 1 0.5 0.2 0.刻度误差02 10容量允差/mL ±0.6%×5 ±1.0%×1 ±1.0%×0.5 ±1.5%×0.2 ±4.0%×0.02 ±0.020不确定度u(v1)/mL 0.017 3 0.005 77 0.002 89 0.001 73 0.000 462 0.011 5温度差异/℃温度波动±4甲醇的膨胀系数/℃-1 1.2×10-3不确定度u(v2)/mL 0.013 9 0.002 77 0.001 39 0.000 554 0.000 055 4 0.027 7相对标准不确定度ur(v) 0.004 44 0.006 40 0.006 41 0.009 08 0.023 3 0.003 00
标准溶液在稀释定容过程中,使用5 mL 移液器1次,1 mL移液器4次(吸取标液1mL3次,500 μL1次),200μL 移液器1次,20μL 移液器1次,10 mL容量瓶7次。则标准溶液配制过程中所使用的玻璃量器和移液器引入的相对标准不确定度为:
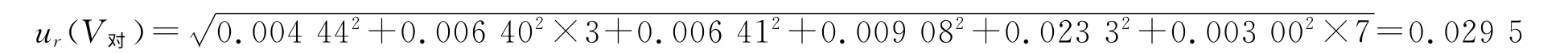
故标准溶液系列配制过程中所引入的相对标准不确定度为:

2.5 仪器引入的不确定度评定(A 类评定)
按照1.2.2实验步骤制备6份平行样品,每份样品莠去津的添加浓度为50μg/kg。测定6份加标样品的莠去津值见表3。

项目次数1 2 3 4 5 6 Xi/(μg/kg) 45.00 45.21 45.52 44.68 44.22 45.97平均值x/(μg/kg) 45.10
单次测量结果的标准偏差为:

因为在实际检测样品时一般只做两个平行样品,所以测量重复性引入的标准不确定度为:

由测量重复性引入的相对标准不确定度:

2.6 回收实验准确度引入的不确定度ur(R)

项目次数1 2 3 4 5 6回 收 率/% 90.0 90.4 91.0 89.4 88.4 91.9平均回收率/% 90.2标准偏差/% 1.223 792
回收实验测定结果见表4,按A 类不确定度,由加标回收率引入的标准不确定度为:

故由加标回收率引入的相对标准不确定度为:

2.7 计算合成不确定度

分量类别 来源 评定方法 分量数值ur(1) 样品制备 B类评定 0.006 59 ur(2) 标准溶液配制 B类评定 0.029 5 ur(xi) 仪器重复性 A 类评定 0.009 67 ur(R) 回收实验 A 类评定 0.005 54
根据不确定度来源分析,可知表5中各分量不确定度各自独立,合成为:
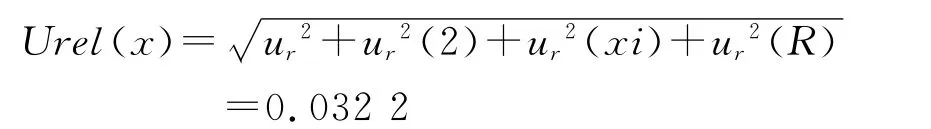
所以
U(x)=0.0322×45.10=1.45μg/kg
2.8 扩展不确定度与测量结果表示
在95%置信水平下,取包含因子k=2,则相对扩展不确定度为:U =2× U(x)=2×1.45=2.90μg/kg。结果表述:该样品中莠去津残留量为45.10μg/kg,则结果表述为(45.10±2.90)μg/kg。
3 结论
通过HPLC-MS/MS外标法测定大米中莠去津残留量的不确定度分析,该测定方法的扩展不确定度为2.90μg/kg。各不确定度分量评定结果显示,标准溶液配制引入的不确定度对合成不确定度的贡献最大。因此,在检测过程中应规范标准溶液配制操作,尽可能减少标准溶液配制步骤降低测定结果的不确定度。
猜你喜欢
中国动物保健(2022年2期)2022-05-05 23:54:04
商品与质量(2021年34期)2021-11-23 21:32:22
云南化工(2020年3期)2020-04-17 03:10:12
技术与市场(2020年5期)2020-03-02 16:06:52
广东茶业(2019年2期)2019-06-18 10:24:24
农药科学与管理(2019年12期)2019-05-20 09:33:26
实验与分析(2018年3期)2019-01-04 03:19:36
实验与分析(2018年4期)2018-02-27 01:20:08
中成药(2018年1期)2018-02-02 07:20:31
福建质量管理(2016年11期)2016-08-16 04:03:04
