
Trace determination and characterization of ginsenosides in rat plasma through magnetic dispersive solid-phase extraction based on core-shell polydopamine-coated magnetic nanoparticles
2020-02-28 11:11NingningZhoShuLiuJunpengXingZifengPiFengruiSongZhiqingLiu
Ningning Zho ,Shu Liu ,Junpeng Xing ,Zifeng Pi ,Fengrui Song ,Zhiqing Liu ,c
a National Center of Mass Spectrometry in Changchun and Jilin Province Key Laboratory of Chinese Medicine Chemistry,Changchun Institute of Applied Chemistry,Chinese Academy of Sciences,Changchun 130022,China
b University of Science and Technology of China,Hefei 230029,China
c State Key Laboratory of Electroanalytiacl Chemistry,Changchun Institute of Applied Chemistry,Chinese Academy of Sciences,Changchun 130022,China
Keywords:
ABSTRACT
1.Introduction
Many traditional Chinese medicines(TCMs),which are homologous to food and medicine,have been widely used in daily food and disease treatment[1].However,their pharmacodynamic material basis or food nutrients remain unclear.The matrix effect of complex biological samples seriously limits the research on active ingredients and metabolites of TCMs[2,3].Thus,trace bioactive constituents and metabolites from biological samples with highly abundant endogenous interference should be enriched to better investigate the pharmacodynamic material basis.At present,the ultra-high-performance liquid chromatography coupled with highresolution mass spectrometry(UPLC-MS)has been widely used to enrich and detect the complex biological samples[4,5].In addition,new acquisition modes that can obtain good data of all precursors and fragment ions for the determination of targeted molecules are developed by the development of data-dependent acquisition(DDA)and data-independent acquisition(MSE)[6,7].Moreover,the analytical sensitivity and capacity are remarkably enhanced[8].Thus,these superiorities have provided an advanced analytical platform to gain comprehensive information for the separation and characterization of metabolites.
Despite the presence of advanced analytical instrumental platforms,there is an urgent need to develop pretreatment methods in conjunction with UPLC-MS for better analysis.At present,traditional solvent extraction methods[9-13],liquid-liquid extraction(LLE)and solid-phase extraction(SPE)[14]are widely used to extract ginsenosides.But these methods require high temperature,long hours,and lots of efforts and solvent.And SPE method needs expensive commercial cartridges and fussy procedures.Although our previously proposed eco-friendly deep eutectic solvent(DES)[2]for extraction of ginsenosides showed high efficiency and solubility,sample concentration is still limited by negligible volatility of DES.The development of magnetic solid-phase extraction(MSPE)and magnetic dispersion solid-phase extraction(MDSPE)[15]has provided a new potential value for the efficient extraction of ginsenosides,but it is a challenge to synthesize functional adsorbents.Table S1 summarizes the advantages and drawbacks of extraction techniques for ginsenosides.
Recently,magnetic nanoparticles(MNPs)have attracted considerable attention due to their outstanding properties,such as good dispersion,controllable core-shell structure,fast and efficient separation,sustainable usage and no need to pack columns.MNPs have been widely used as adsorbents for enriching small molecules[16,17].However,MNPs cannot form other interactions except for hydrogen bonds between most TCMs or food bioactive constituents due to the lack of specific functional groups.Moreover,the syntheses of materials require complex procedures and harmful solvents in the preparation process,and few are applied to complex biological samples.
Dopamine(DA),a small molecule that mimics the adhesive proteins of mussels,has been confirmed to play a unique role in the interfacial adhesion of numerous material surfaces by selfpolymerization under sufficient mild conditions(weak alkaline medium)[18-20].For example,self-polymerization of DA has been used for the extraction of estrogenic compounds from cow,goat,sheep and human milk[21].Unfortunately,the reported core-shellbased polydopamine NPs(PDA NPs)still have some limitations.The first limitation is that PDA possesses strong interfacial adhesion,which results in blocking cavities of buried substance in molecular imprinted materials(MIPs).Moreover,few studies report the detection of targeted molecules from complex matrix samples.
In this study,a novel strategy was developed to enrich and detect ginsenosides from complex biological samples by introducing UNIFITWlibraries assisted with Fe3O4@SiO2@PDA NPs as adsorbents of MDSPE.The strategy incorporated the advantages of the three approaches:(1)MDSPE with Fe3O4@SiO2@PDA NPs for fast and efficiently capturing trace bioactive constituents from biological samples without packing column;(2)UPLC-MS technology for fast and accurate detection of trace constituents;(3)Supplemental UNIFI libraries for fast,high-throughput and automatic match of data and identification of compounds.This strategy achieved a promote platform including pre-treatment,sample analysis based on MS and data analysis.For experiments,the conditions were statistically optimized by response surface methodology(RSM)with total ginsenosides to obtain the best extraction process.Subsequently,the strategy was applied for the fast detection and determination of metabolites from rat plasma,which eliminated the interference of several endogenous substances.Moreover,the synergetic strategy was accomplished in compliance to 9 of the 12 green chemistry principles with green analytical chemistry's(GAC)[22].The proposed method was proven to be a promising strategy to improve the enrichment capability and sensitivity of MS for tracing the targeted molecules in biological samples.
2.Experimental
2.1.Chemicals
White ginseng(WG),total ginsenosides(extract of WG)and all the standards of ginsenosides Rb1,Rb2,Rb3,Rc,Rd,Re,Rg1,PPD,Rf,Rg2,Rg3,Rh1,Rh2 and compound K were obtained from the College of Pharmacy,Jilin University.Dioscin as an internal standard(IS)was purchased from National Institutes for Food and Drug Control(Beijing,China).HPLC-grade acetonitrile and methanol were acquired from Fisher Scientific(Loughborough,UK).Ultrapure water was prepared by a Milli-Q water(18.2 MΩ)purification system(Milford,MA,USA).Tris(hydroxyl methyl amino methane)(Tris),dopamine hydrochloride(DA·HCl),heparin,ammonium sulfate(NH4)2SO4,aminopropyl triethoxy silane(APTES),tetraethoxysilane(TEOS)and Fe3O4nanoparticles(20-30 nm)were obtained from Aladdin Industrial Corporation(Shanghai,China).HCl,acetic acid and methanol were provided by Sinopharm Chemical Reagent Co(Shenyang,China).A 5 mM Tris-HCl buffer solution(pH 7-9)was prepared by us.
2.2.Instrumentations
The morphology and Energy Dispersive X-Ray(EDX)spectroscopies of MNPs were obtained by the scanning electron microscopy(SEM,JSM-6460LV,Japan).Fourier transform infrared spectroscopy(FT-IR,IFS 66 V/S,Germany)was performed to identify the materials with KBr.Other characteristics were measured by vibrating sample magnetometry(VSM,MPMS XL,USA)at room temperature and a D8 advance X-ray diffractometer(XRD,D8,Germany)with Cu Kαradiation(λ=1.5406Å).The UPLC-MS/MS analyses were performed by a Waters ACQUITY UPLC system(Waters Corp.,Milford,USA)coupled to a triple quadrupole(TQ)mass spectrometer equipped with the electrospray ionization(ESI)source in multiple reactions monitoring(MRM)mode.A Q-TOF SYNAPT G2-S High Definition Mass Spectrometer coupled to ESI(Waters Crop.,Manchester,UK)was adopted for detection of ginsenosides metabolites before and after treatment with Fe3O4@SiO2@PDA NPs.The C18 column(50 mm×2.1 mm,1.9μm;Thermo scientific)was employed in the entire experiments.MassLynxV4.1 and Marker-Lynx Application Manager(Waters Corporation,Milford,USA)and UNIFI 1.8(Waters,USA)were carried out to process the collected data.And all detailed steps of mass spectrometry analyses were described in Supplementary data(Methods section),including UPLC-MS/MS analysis,Q-TOF analysis and mass spectral data analysis with UNIFITW.
2.3.Synthesis of the Fe3O4@SiO2,Fe3O4@SiO2@APTES and Fe3O4@SiO2@PDA NPs
The Fe3O4@SiO2NPs were synthesized by the previous method[23].The Fe3O4@SiO2@APTES NPs were synthesized through the modified method[24].The synthesis strategy of Fe3O4@SiO2@PDA NPs based on the modified method[25]through one-pot synthesis is as follows:briefly,the Fe3O4NPs(100 mg)were dispersed into an ethanol solution(95% ethanol:water,v/v).Then,ammonium hydroxide was added into the mixture solvents(pH=8.5).Ultrasound 10 min later,0.2 mL of TEOS was added dropwise into the system for 6 h at 40℃.After that,90 mg of DA·HCl was added into the bottle,followed by stirring for 1 h.Subsequently,the obtained product was isolated by the external magnetic field and washed by water and ethanol.Eventually,the product was dried under vacuum at 40℃ for 24 h.
2.4.Animals
Six male rats,weighting 200±20 g,were obtained by Jilin Medical University(Jilin,China).After a week of adjustable feeding,they were administered WG(7 g/kg)once a day for 5 days.The rats were sacrificed 2 h after the last gavage,and the blood was collected and centrifuged.The supernatants were stored at-80℃.All animals’experiments and the procedures followed the Guide for the Care and the Use of Laboratory Animals of Jilin University.
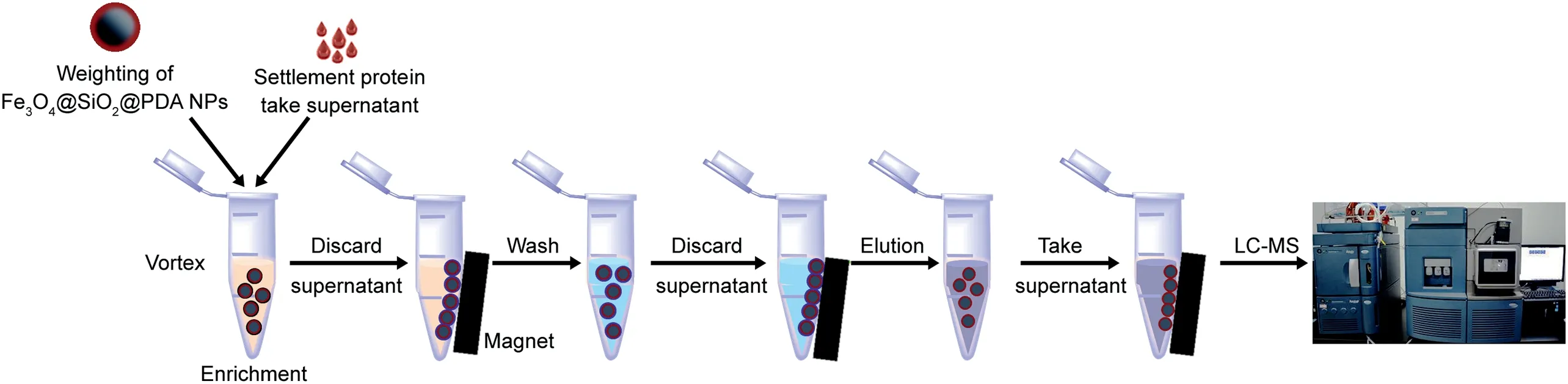
Scheme 1.Schematic presentation of the proposed strategy for capture and characterization of ginsenosides by Fe3O4@SiO2@PDA NPs based on UPLC-MS method.
2.5.Enrichment of ginsenosides from total ginsenosides
During MNPs screening and tailoring,5 mg of the MNPs were mixed with 1 mL of the total ginsenosides in water(500μg/mL).The mixture was vortexed and shaken for 2 h.Later,the MNPs were isolated by external magnetic field,followed by ultrasound-assisted cleaning with 1 mL of methanol-acetic acid(9:1,v/v)and then direct mass spectrometry analysis.
The adsorption capacity of Fe3O4@SiO2@PDA NPs for ginsenosides was investigated at different concentrations of total ginsenosides while other conditions remained unchanged.Meanwhile,the dynamic adsorption capacity was evaluated by varied time from 10 min to 360 min.Desorption time was estimated at varied time from 10 min to 120 min.The adsorption amounts(Q,mg/g)of MNPs were calculated according to Eq(1)[26].

where,Co(μg/mL),Ce(μg/mL),V(mL)and m(g)represent the initial and equilibrium concentration of ginsenosides,the volume of the total ginsenosides solutions and the mass of the MNPs,respectively.
To investigate the maximum adsorption properties of Fe3O4@-SiO2@PDA NPs,the adsorption isotherm was calculated by using a series of total ginsenosides solutions with different concentrations.The kinetic tests were calculated by varied different adsorption time.The Langmuir and Freundlich isotherm models,and the pseudo-first-order and pseudo-second-order models were adopted to evaluate the equilibrium and kinetic data,respectively[27].
2.6.Experimental design and statistical analysis for total ginsenosides
The screening experiments,including synthetic species of magnetic materials,content of dopamine,pH of the solvent system,concentrations of the total ginsenosides,enrichment time and desorption time were carried out.According to the above obtained result of single factor experiments,the appropriate variable range in extraction process(shown in Table S2)was employed for experimental design using RSM.The Box-behnken design(BBD)combined with RSM was performed as follows:pH(7-9),concentrations of the total ginsenosides(100-700μg/mL),enrichment time(30-80 min),and elution time(10-80 min)at levels(-1,0,1).Here,the reason for choosing such pH range was that the extraction efficiency was better at weak alkalinity for ginsenosides[15].The extraction recoveries of ginsenosides in 29 experiments(three replicates)were estimated by BBD in Table S3.
Design-Expert 8.0.6 software[28]was operated to evaluate the statistical significance in design experiment.Based on analysis of variance(ANOVA),the quadratic multiple regression models were built to investigate model quality according to the square of correlation coefficient(R2)and the lack of fit which evaluated the failure of the model.
2.7.Real samples extraction procedure

Fig.1.SEM images of Fe3O4,Fe3O4@SiO2,Fe3O4@SiO2@APTES and Fe3O4@SiO2@PDA NPs.
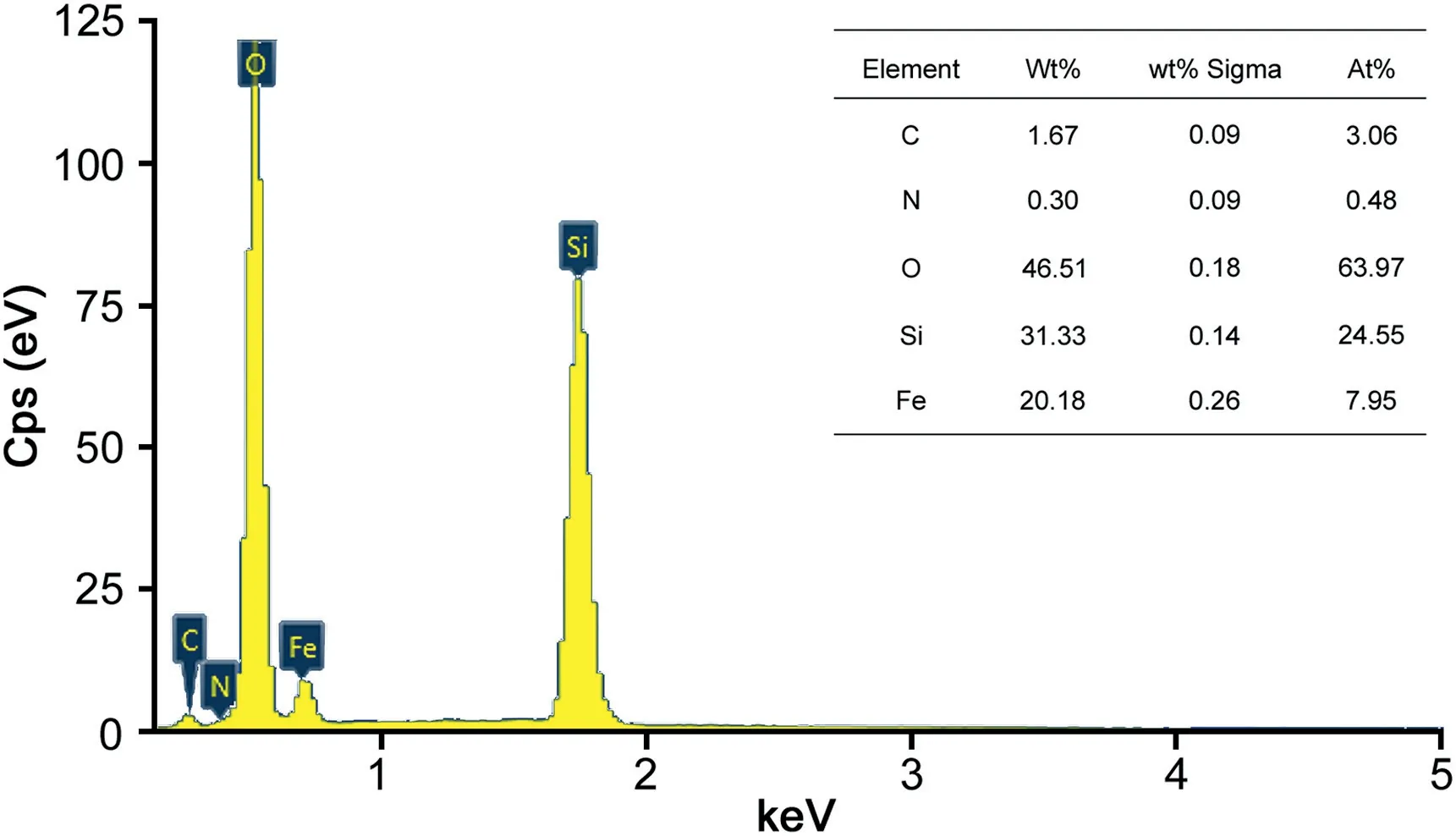
Fig.2.The EDX spectrum and element compositions of Fe3O4@SiO2@PDA NPs.
The real plasma samples after lavage WG were analyzed by our synergetic strategy in Scheme 1.The 200μL plasma samples were divided into conventional methanol group and Fe3O4@SiO2@PDA NPs enrichment group(six replicates).First,all plasma samples were dealt with 3 folds methanol.After centrifugation,the supernatants were dried under nitrogen in methanol group.However,in the PDA group,5 mg Fe3O4@SiO2@PDA NPs were added into 1 mL Tris-HCl buffer system containing the supernatants of the same volume(final pH=8.78).After vortexing for 2 min,the system was shaken for 80 min.Later,the obtained supernatant was removed by external magnetic field.And the MNPs were eluted with 1 mL of methanol-acetic acid(9:1,v/v)by ultrasound for 68.25 min.Finally,the eluted solvents were also dried under nitrogen.All both dried samples were re-dissolved with 50μL methanol for TOF analysis.
2.8.Method validation and measurement uncertainty
A series of working solutions of Rb1,Rb2,Rb3,Rc,Rd,Re,Rg1,PPD,Rf,Rg2,Rg3,Rh1,Rh2 and compound K were obtained by stepwise dilution of the stock solutions in methanol.The calibration was created in the form of y=ax+b.The limits of detection(LODs)and limits of quantification(LOQs)were recognized at a signal-to-noise ratio(S/N)of 3 and 10 times.For quality control(QC)samples,fourteen ginsenosides were operated at low,medium,and high concentrations in WG powder and plasma,respectively.The IS was prepared at a concentration of 0.1μg/mL in each sample.Intra-and inter-day accuracy and precision were assessed by QC samples at concentration levels of low,middle and high for 14 ginsenosides in 3 days.The relative standard deviation(RSD)and relative error(RE)represented the precision and accuracy,respectively.To investigate the matrix effect,the analyses of total ginsenosides and drug-free plasma samples spiked with the 14 analytes were carried out.To further estimate the extraction recovery,the samples of WG powder and drug-free plasma samples spiked with the 14 analytes were directly analyzed using our proposed method.
Proper quality assurance/quality control(QA/QC)procedures are vital for the measurement results,which provide reliable analytical information.According to Konieczka and Namie′snik[29],amount of sample,analyte concentration,calibration step,repeatability of results and recovery determination(trueness)of the uncertainty components were estimated.The uncertainty of results was calculated based on Karimi et al.[30]and Borecka et al.[31].Expanded uncertainty was determined by the Eq(2).
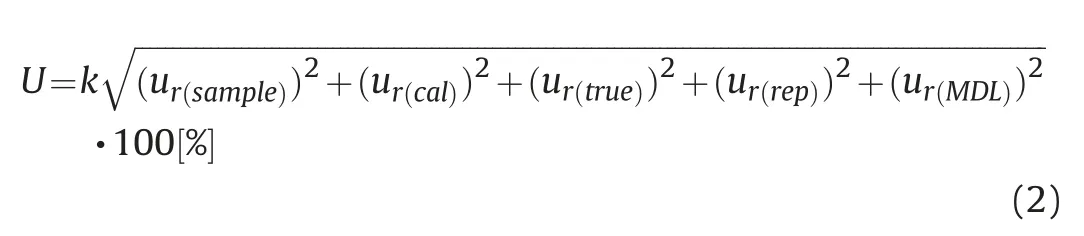
The meaning of each element in the Eq(2)is as follows:U-expanded uncertainty(%),k-coverage factor(k=2 for P=95%),ur(sample)-relative standard uncertainty of sample,ur(cal)-relative standard uncertainty of calibration step,ur(true)-relative standard uncertainty of trueness,ur(rep)-relative standard uncertainty of repeatability,and ur(MDL)-relative standard uncertainty of MDL determination.Meanwhile,ur(cal),ur(true),ur(rep)and ur(MDL)were calculated by the following Eqs(3)-(6),respectively.
The sensitivity of the method is expressed by method detection limit(MDL)and method quantitation limit(MQL)for the entire procedure.And the MDL and MQL were calculated by the Eq(7)and Eq(8).


Fig.3.(A)X-Ray diffraction patterns of Fe3O4,Fe3O4@SiO2,Fe3O4@SiO2@APTES and Fe3O4@SiO2@PDA NPs.(B)FT-IR spectra of all the NPs.(C)VSM magnetization curves of all the NPs.(D)VSM magnetization curves of synthetic Fe3O4@SiO2@PDA NPs at different reaction times.
3.Results and discussions

where SDxyis the standard deviation of the calibration curve,p is the repetition number of measurements for a given sample,m is the total number of standard samples used for plotting the calibration curve,Xsampleis the concentration of sample,Xmis the mean value of all concentrations of a standard solution for which the measurement was made to plot a calibration curve,Xiis the calculated concentration,b is the slope of the calibration curve,RSDRis the RSD derived from the recovery,RSDresultsis the RSD in real samples and Cmis the mean concentration of analyte.The analytical result is expressed as mean±expanded uncertainty.
3.1.Characterization of the magnetic nanoparticles
3.1.1.SEM and EDX characterizations
All MNPs were characterized through SEM.As shown in Fig.1,the diameters of Fe3O4,Fe3O4@SiO2,Fe3O4@SiO2@PDA and Fe3O4@SiO2@APTES NPs were about 25 nm,130 nm,180 nm,and 220 nm,respectively.The results revealed that the size of particles increased with the layer-by-layer modification on the Fe3O4core,and the order is expressed as follows:Fe3O4NPs<Fe3O4@SiO2 NPs<Fe3O4@SiO2@PDA NPs<Fe3O4@SiO2@APTES NPs.In addition,the enrichment capability of different MNPs for ginsenosides was also estimated through MS analysis(MRM conditions,calibration curves and analytical results are shown in Tables S4 and S5 and Fig.S1).The order of enrichment capability was in consistent with the order of MNPs size.Although the specific surface areas of Fe3O4and Fe3O4@SiO2NPs were larger than the other two,the enrichment capabilities of bare MNPs and hydroxyl-containing Fe3O4@-SiO2NPs were weaker than the other two amino-containing MNPs.It is obvious that the effect of amino groups was greater than hydroxyl groups,and the effect of functional groups was relatively more important than specific surface area in this experiment.Moreover,the higher enrichment capability for Fe3O4@SiO2@PDA NPs compared with Fe3O4@SiO2@APTES NPs was obtained due to two reasons:(1)PDA possessed multiple recognition sites,such as amino and hydroxyl groups;(2)Fe3O4@SiO2@PDA NPs with small size had relatively high surface areas.

Fig.4.RSM response surface plots of the model for extraction of ginsenosides.
Furthermore,the EDX spectrum and main elemental composition analysis for Fe3O4@SiO2@PDA NPs are presented in Fig.2.The contents of N elements were further validated the existence of PDA.All the results suggested that PDA was successfully grafted to the Fe3O4@SiO2NPs.
3.1.2.XRD characterization
The XRD patterns are shown in Fig.3A.It was obviously observed that six major characteristic diffraction peaks of Fe3O4NPs appeared at 2θ=30.2°(220),35.5°(311),43.2°(400),53.4°(422),57.2°(511),and 62.9°(440).Although the Fe3O4NPs were gradually modified,the spectra of modified MNPs were similar to the XRD spectrum of Fe3O4NPs at the six major peaks,revealing that the modification had not been destroyed on the crystal structure of Fe3O4particles.The peaks attributed to SiO2and PDA were not observed,because SiO2or PDA coating on the surface of Fe3O4mainly existed as amorphous forms[32].
3.1.3.FT-IR characterization
The Fe3O4,Fe3O4@SiO2,Fe3O4@SiO2@APTES and Fe3O4@-SiO2@PDA NPs were further validated by FT-IR(Fig.3B).The typical peaks located at approximately 576 cm-1and 1090 cm-1were attributed to the stretching vibration of Fe-O and Si-O bonds,respectively[33].The peak at approximately 1626 cm-1was assigned to the N-H vibration in APTES[34].The peaks for PDA at approximately 1621 cm-1and 1508 cm-1 were assigned to C=C stretching vibrations in the aromatic ring and N-H bending vibration,respectively[35].Moreover,in the entire spectrum,a broad band at approximately 3420 cm-1corresponded to the-NH or-OH stretching vibration[32].These results reaffirmed the formation of PDA shell on Fe3O4@SiO2@PDA NPs.
3.1.4.VSM analysis
As observed in Fig.3C,the saturation magnetizations of Fe3O4and Fe3O4@SiO2NPs were 53.08 emu/g and 42.40 emu/g,which reduced to 33.07 and 37.71 emu/g after coating with APTES and PDA,respectively.Although the saturation magnetization declined clearly,the MNPs still met the experimental requirements.The saturation magnetization of Fe3O4@SiO2@PDA NPs was stronger than that of Fe3O4@SiO2@APTES,which proved its efficient separation in the adsorption of ginsenosides.As shown in Fig.3D,saturation magnetization reduced with the increase of polymerization time.And the intensity was the highest when the polymerization time was 60 min.This optical condition demonstrated the existence of critical values for polymerization time.
3.2.Effect of content of DA
The content of DA was estimated for the synthesis of Fe3O4@-SiO2@PDA NPs to improve their enrichment capability(Q).As shown in Fig.S2A,Q was the strongest when DA·HCl was 90 mg.Excess amounts of DA·HCl caused the decrease of extraction capacity for ginsenosides.The results showed that superfluous DA·HCl promoted self-aggregation of DA,or the shell of MNPs thickened and resulted in the decrease of specific surface area[36].
3.3.Effect of reaction time
The polymerization time was also estimated for the synthesis of Fe3O4@SiO2@PDA NPs.As displayed in Fig.S2B,Fe3O4@SiO2@PDA NPs exhibited the highest extraction efficiencies of ginsenosides when the polymerization time was 60 min,which resulted from the increasement of recognition sites,a stepwise suitable thickness andthe above-mentioned strong saturation magnetization.However,the enrichment effect became relatively low with long polymerization time.The low enrichment effect was caused by the reduction of magnetism and specific surface area due to the thickened shell of the MNPs as time went by,and this finding was consistent with the characterization data in Fig.3D.
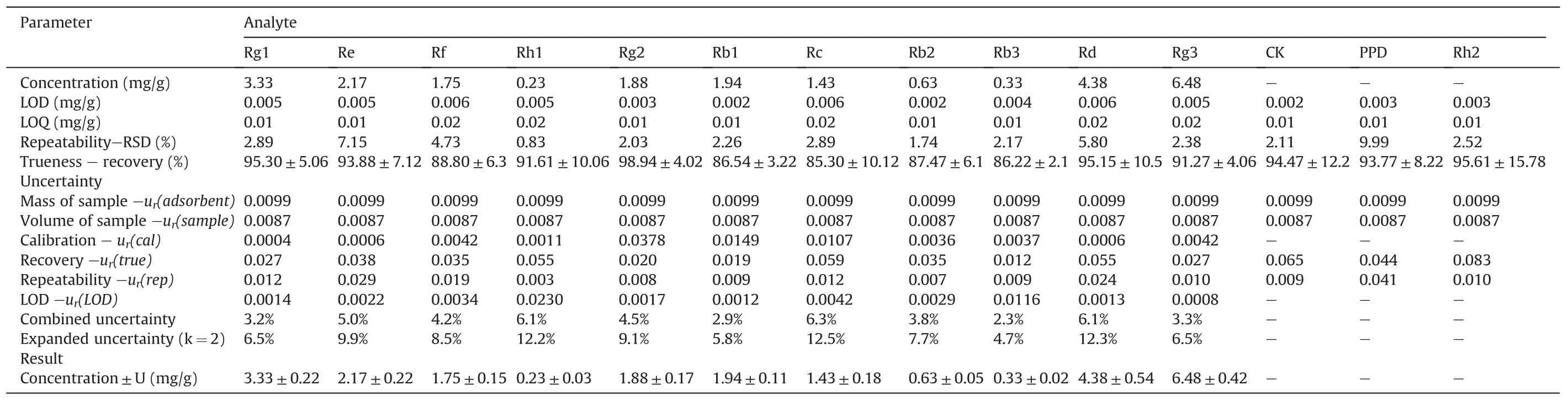
Table 1Calculated values of relative standard uncertainties,combined standard uncertainties and expanded uncertainties for the determination of 14 analytes in real plasma.
3.4.Optimization for ginsenosides extraction using RSM
Single-factor experiments were implemented to obtain the suitable value ranges for statistical analysis(Fig.S3).In Fig.S3A,when pH ranged from 7 to 9,the values of adsorption capacities increased.It is confirmed that the adsorption capacities for ginsenosides were better at weak alkalinity for the following two reasons:(1)ginsenosides formed more hydrogen bonds with the multiple recognition sites of PDA at weak alkalinity;(2)hydrophobic interaction was better at weak alkalinity.For the isotherm test,Langmuir model(R2=0.94)was more successful and suitable than Freundlich model(R2=0.89)at 10-1000μg/mL(Total ginsenosides solutions),and the fitting Q was greater by Langmuir model(Fig.S3B).For the adsorption kinetic test(Fig.S3C),the Q was coincided with the pseudo-first-order model,and the pseudo-firstorder model(R2=0.99)was more suitable than pseudo-secondorder model(R2=0.86).As shown in Fig.S3D,the desorption capacity remained unchanged after about 60 min.All those results suggested that the single-factor experiment could provide certain ranges for RSM analysis.Furthermore,Design-Expert 8.0.6 was used to evaluate the statistical relevance in the suitable range(Fig.4).Second-order polynomial quadratic equations between extraction yields(Y)and coded variables(A,B,C and D)were calculated and expressed as follows:

The data of ANOVA analysis and 3D response plots indicated the relevant variables for Y of ginsenosides and the interaction effect between the variables(Table S6).In the model,extraction concentrations(B),enrichment time(C),and elution time(D)exhibited significant difference at p<0.05.pH displayed an insignificant efficiency(p=0.0856).The results indicated that B,C and D presented remarkable effects,and no obvious interactions with each other were observed during the enrichment process.A predicted optimal Y of 29.63 mg/g was obtained when the extraction system pH was 8.78,concentration was 576.32μg/mL,enrichment time was 80 min,and elution time was 68.25 min.Meanwhile,the highest Yof 28.32±0.82 mg/g was achieved and the result reflected the repeatability of the model obtained.Generally,a good methodological result was obtained.In addition,the recovery exceeded 80%after recycling Fe3O4@SiO2@PDA NPs 6 times(data are shown in Fig.S4),and this finding revealed that the MNPs were efficient and sustainable for the enrichment of ginsenosides.
3.5.Application to plasma samples and metabolite identification
From BBD experiments,the best extraction process was optimized and the recovery was extremely close to predicted values.The LODs and LOQs of analytes were obtained through the verification of methodology,and good linearities were higher than 0.99 and wide linear ranges were obtained.In Table S7,the accuracy and precision of all the analytes were evaluated.All values conformed to methodological standard.In Table S8,mean accuracies of extraction recoveries and matrix effects of all the analytes ranged from 85%to 115% at three QC levels(n=6).The high extraction recovery and less matrix effect of 14 analytes indicated that the Fe3O4@SiO2@PDA NPs extraction method was viable.The method was feasible when ginsenosides were enriched from blank plasma spiked with standard mixtures of ginsenosides(in methanol)by the Fe3O4@-SiO2@PDA NPs.In addition,the QA/QC procedures were successfully applied to the measurement and analytical results.Finally,uncertainties of all measurement and analytical results were calculated and the final expanded uncertainty results are shown in Table 1.To further evaluate whether the proposed method has high sensitivity for Q-TOF MS analysis in real complex samples,the proposed approach was implemented to the rat plasmaadministered WG,and UNIFI™libraries were supplemented to automatically assist the separation and characterization of compounds.The base peak ion(BPI)chromatograms of conventional methanol group and Fe3O4@SiO2@PDA NPs enrichment group are summarized in Fig.5.As shown in BPI chromatograms,ion suppressions from plasma had a severe effect on the mass spectrum of ginsenosides,although the signal intensity was slightly high for the conventional methanol method.
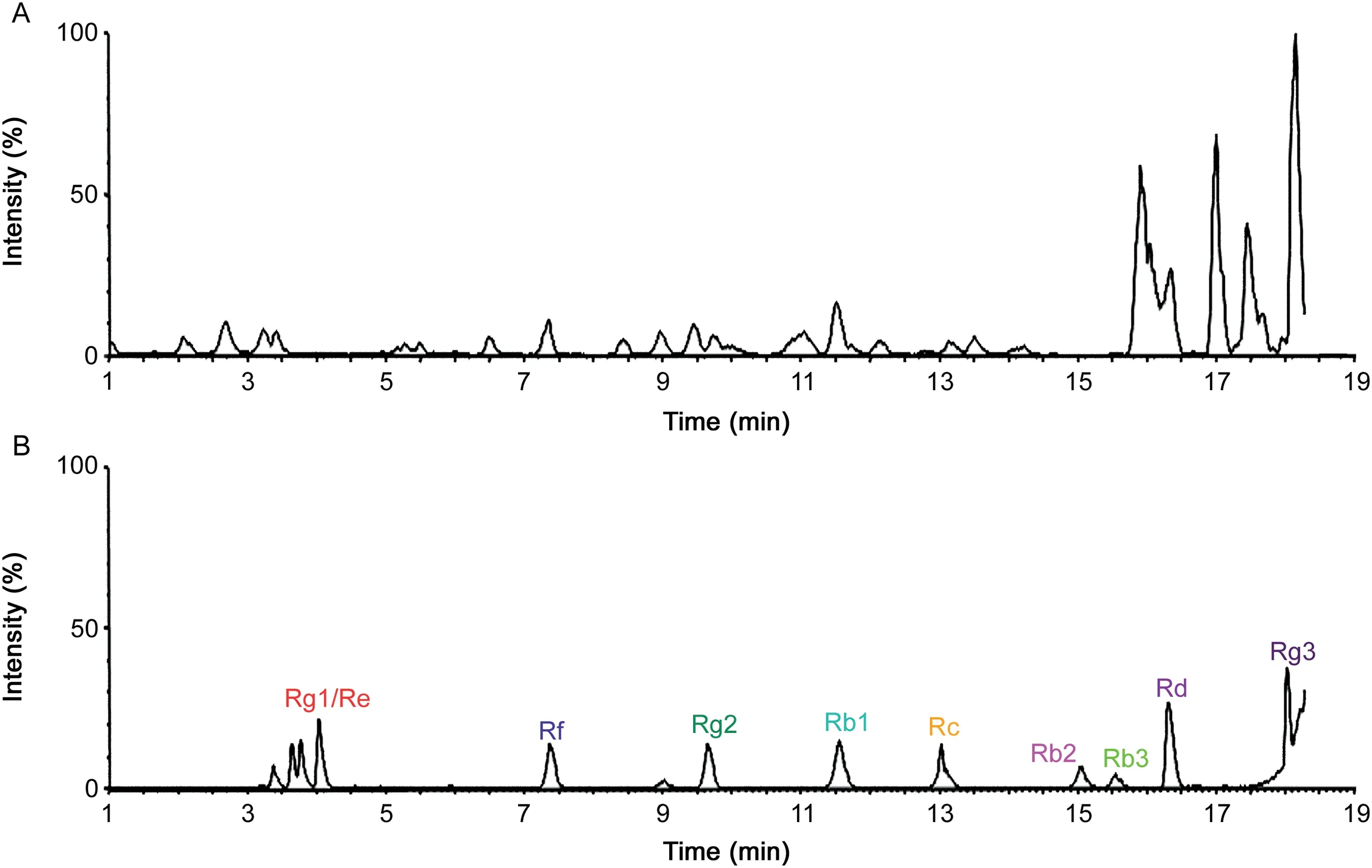
Fig.5.The BPI chromatograms acquired by conventional methanol extraction method(A)and Fe3O4@SiO2@PDA NPs enrichment method(B).
Compound 3(recognized as ginsenosides Re)was taken as examples to illustrate the structural identification of ginsenosides based on mass spectral fragmentation pathways[15]and UNIFI™libraries.As shown in Fig.6,characteristic ions of compound 3 were m/z 945.5379[M-H]-and 991.5471[M+HCOOH-H]-in the negative ion mode.For the high-energy function,the main fragmentation ions were at m/z 799.4739,783.4918,637.4239,475.3763,391.2855 and 161.0451,which corresponded to[M-Rha]-,[M-Glc]-,[M-Glc-Rha]-,[M-Glc-Rha-Glc]-,[M-Glc-Rha-Glc-C6H12]-and[C6H9O5]-,respectively.These fragmentation ions were identical to those of Re in the UNIFI™libraries.Moreover,the liquid retention time of compound 3 was the same as the Re in UNIFI™libraries.So,compound 3 was identified to ginsenoside Re through constant comparison and confirmation.All the prototypes and metabolites in vivo were matched through the above characterization and confirmation steps,the results are listed in Table 2.The combination of MSEand UNIFI showed a huge potential in the separation and determination of compounds.
3.6.Comparison of the proposed method with other methods
Table S9 shows the distribution information of each ginsenoside in the biological samples detected by different enrichment methods.Surprisingly,23 compounds were detected and determined using our proposed method,whereas only 8 ginsenosides were determined by the methanol method.For the proposed enrichment method,the ion signal enhancement effect of targeted compounds was obvious.Moreover,the proposed strategy can rapidly separate and identify many targets in the spectrum,which avoided several endogenous substances.This finding proved that Fe3O4@SiO2@PDA NPs had a huge potential for the enrichment of ginsenosides from a wide range of samples.
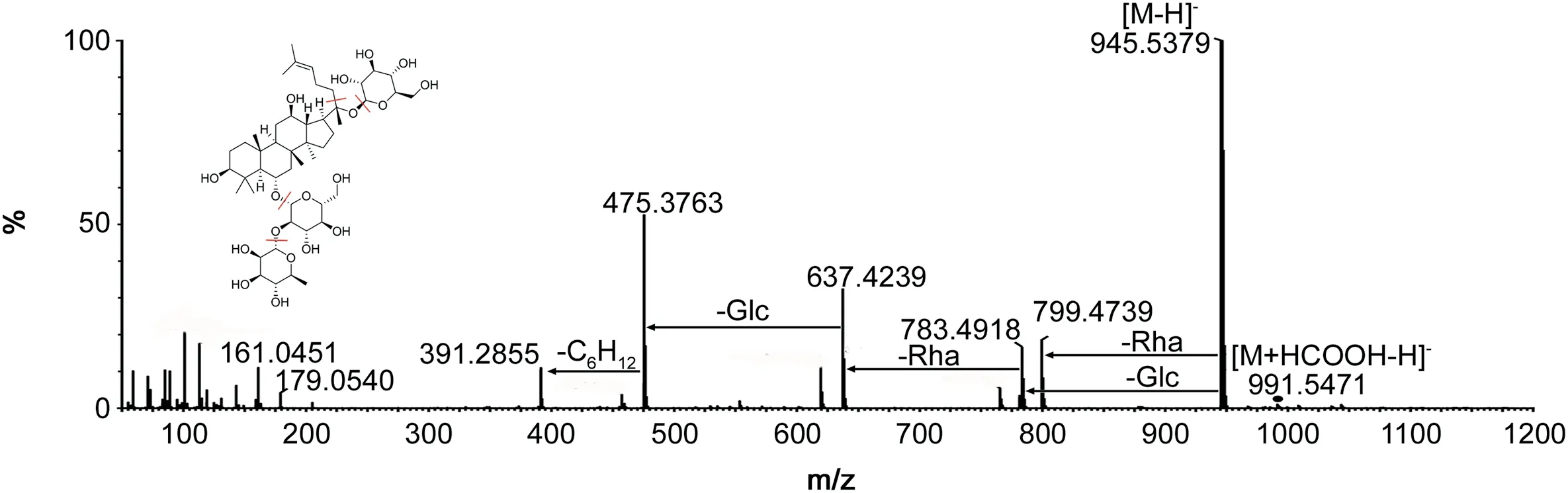
Fig.6.The major fragmentation pathways of ginsenoside Re.
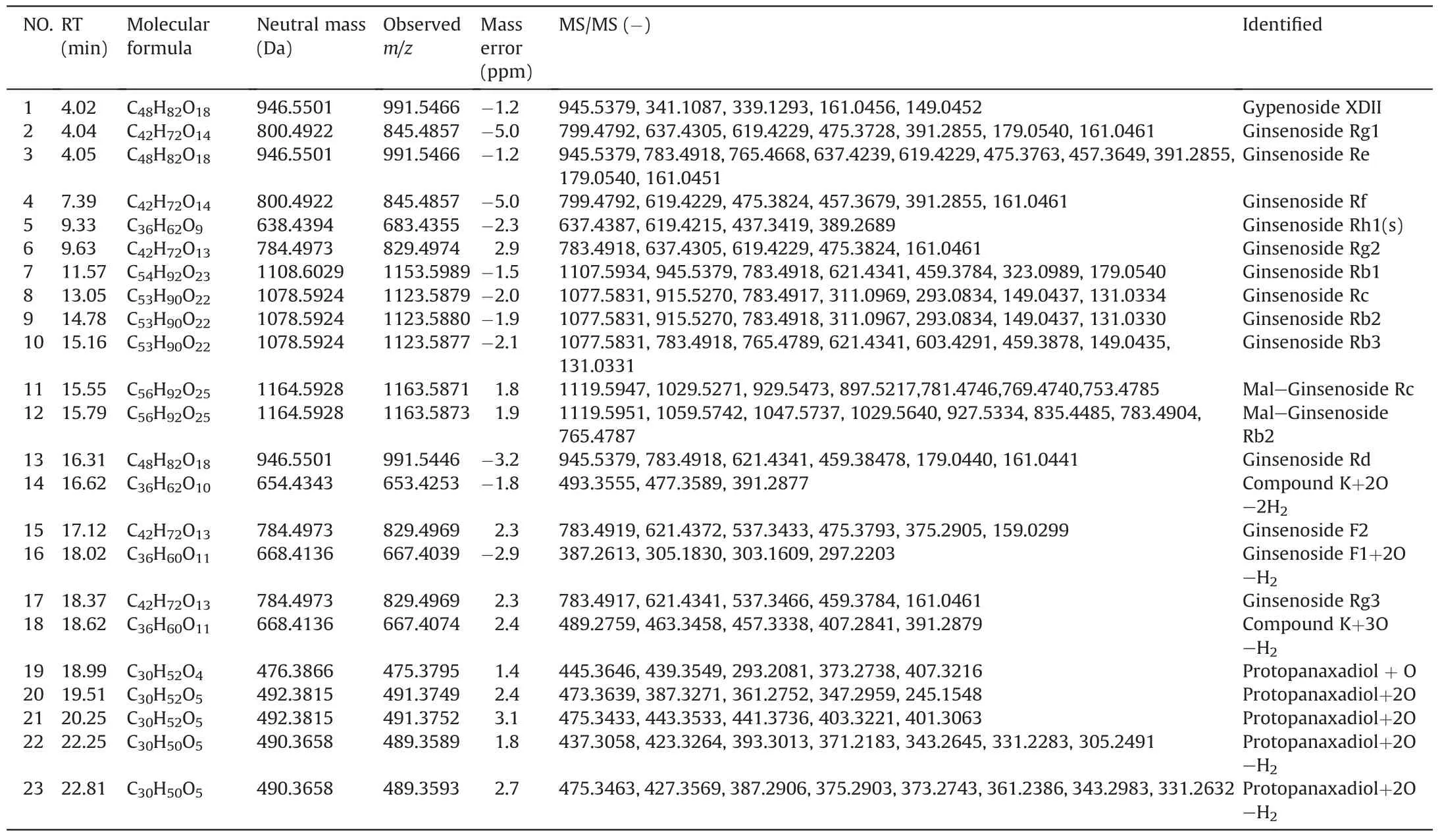
Table 2The major prototypes and metabolites of enrichment and identification using Fe3O4@SiO2@PDA NPs extraction method in the biological samples in the negative ion mode.
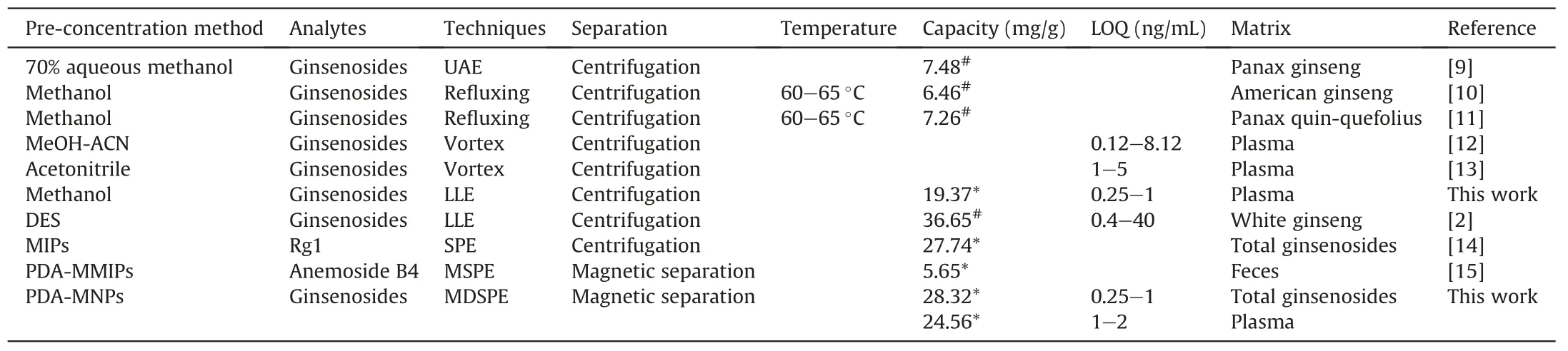
Table 3Comparison of adsorption capability of pre-concentration methods for determination of ginsenosides.
As shown in Table 3,the proposed strategy was compared with previous extraction methods for the determination of ginsenosides[2,9-15].The proposed strategy had several advantages over normal methods.Compared with conventional solvent extraction and LLE,MDSPE did not require centrifugation and can perform rapid separation.Moreover,the adsorption capability of Fe3O4@-SiO2@PDA NPs for ginsenosides was the highest(28.32 mg/g in total ginseng and 24.56 mg/g in plasma).All these results demonstrated that the proposed method was a fast,sensitive and reusable technique that could be used for the enrichment and determination of ginsenosides in biological samples.
As for green aspects,the above-mentioned techniques for the enrichment of ginsenosides are comprehensively assessed in Table S10.The proposed strategy was accomplished in compliance to 9 of the 12 GAC principles,which stood for reduction of the use of chemical substances and the recycle of them.All other methods met no more requirments of the principles than the one used in this work.
4.Conclusions
In summary,Fe3O4@SiO2@PDA NPs with multiple recognition sites in PDA were synthesized through a green one-pot procedure.It is easy to synthesize and there is no need to pack columns for sample analysis.Importantly,the materials could recycle at least 6 times,conforming to the GAC principles.Moreover,the fast separation and characterization of 23 ginsenoside prototypes and metabolites from plasma were achieved by combining UPLC-MS with UNIFI libraries.In comparison,conventional methanol method can only detect 8 ginsenosides from the same plasma samples.This new synergetic strategy for the accurate characterization of trace ginsenosides using an easy,fast,economical,automated and ecofriendly way shows potential applications.The integration of facile and one-pot materials,advanced analytical platforms and automatically high-throughput post data-processing softwares will promote the research on the pharmacodynamic material basis of related food and TCMs.
 Journal of Pharmaceutical Analysis2020年1期
Journal of Pharmaceutical Analysis2020年1期
- Journal of Pharmaceutical Analysis的其它文章
- Karacoline,identified by network pharmacology,reduces degradation of the extracellular matrix in intervertebral disc degeneration via the NF-κB signaling pathway
- Nanodiamonds with powerful ability for drug delivery and biomedical applications:Recent updates on in vivo study and patents
- Comparing different domains of analysis for the characterisation of N-glycans on monoclonal antibodies
- Rapid identification of chemical profile in Gandou decoction by UPLCQ-TOF-MSE coupled with novel informatics UNIFI platform
- GC-NICI-MS analysis of acetazolamide and other sulfonamide(R-SO2-NH2)drugs as pentafluorobenzyl derivatives[R-SO2-N(PFB)2]and quantification of pharmacological acetazolamide in human urine
- Analysis of pesticide residues in commercially available chenpi using a modified QuEChERS method and GC-MS/MS determination