
不同制备方法对Cu-Co/ACN同时脱除Hg0和CO性能的影响
2020-02-25 00:45唐晓龙王雨禾易红宏高凤雨
中南大学学报(自然科学版) 2020年1期
唐晓龙,王雨禾,易红宏,高凤雨
(1.北京科技大学能源与环境工程学院,北京,100083;2.工业典型污染物资源化处理北京市重点实验室,北京,100083)
烧结工艺是钢铁工业的重要组成部分,烟气波动性大,组成复杂。据统计,烧结工艺排放钢铁工业约41%的Hg[1],其总Hg 排放系数为1.28~2.49 mg/t[2]。此外,烧结烟气含有较高体积分数的CO,为0.5%~1.0%。CO可以与血红蛋白结合,抑制人体呼吸作用,对环境和人体造成了严重危害。Hg 是1 种具备挥发性、高毒性和生物累积性的重金属污染物[3],元素汞(Hg0)、氧化汞(Hg2+)和颗粒汞(Hgp)是烧结烟气中Hg的主要存在形式[4],其中,Hg2+和Hgp可以被现有烟气净化系统有效脱除[5],Hg0具有较强稳定性和较低可溶性,是烟气脱Hg研究重点[6]。Hg0被催化氧化为Hg2+后,可吸附性提高,最终实现Hg0的有效脱除。SCR脱硝催化剂在300~400 ℃具备Hg0脱除能力[7],但并不适用于烧结烟气(80~200 ℃)中Hg0的脱除。除贵金属(如Ir[7])外,过渡金属Fe[8],Co[9],Ce[10],Mn[11-12]和Cu[6]均对Hg0具备一定的低温催化氧化活性。活性碳和活性焦[6]等材料具有比表面积高、机械强度高和易于改性等优点,通常被用作催化剂载体。活性焦的金属改性可以显著提升其脱Hg0能力,是实现烧结烟气脱除Hg 的有效手段。随着工业烟气CO 排放量增加,CO的催化氧化脱除成为学者研究的重点。贵金属催化材料在低温条件下可以实现高效CO 催化氧化[13-14],但其制备成本较高,不利于大规模应用。采用Cu 和Mn 复合金属材料制备的Hopcalite 催化剂被广泛应用于CO 催化氧化[15]。金属材料表面掺杂Co[16]或Ce[17-18]可以有效提高其低温CO催化氧化的性能。综上所述,过渡金属元素Co 和Cu 等具备脱Hg0能力,同时也具备催化氧化CO的能力,这为开发双功能催化材料提供了研究依据。本文以硝酸预处理的活性焦(ACN)为载体,首先,考察不同负载量的Co以及掺杂Cu后对活性焦催化剂催化性能的影响;其次,以Cu和Co为活性组分,分别采用等体积浸渍法(VI)、沉淀-沉积法(DP)、水热合成法(HY)和溶胶-凝胶法(SG),制备系列Cu-Co/ACN催化剂,考察其同时脱Hg0脱CO 的性能;最后,通过BET,XRD,ICP,H2-TPR 和FTIR 分析上述4 种方法制备的催化剂表现出性能差异的本质原因。
1 材料与方法
1.1 活性焦载体的预处理
柱状活性焦(AC)购自巩义市北山口活性炭厂。
1)筛分清洗。将活性焦进行碾磨,用筛子筛分出粒径为0.250~0.425 mm 的颗粒,用去离子水清洗4~5 次,最后,置于110 ℃的烘箱中干燥12 h,取出备用。
2)预处理。将备用的活性焦(20 mL/g 活性焦)用质量分数为30%硝酸加热煮沸1 h,冷却后用去离子水洗至中性,最后,置于110 ℃下干燥12 h,所得颗粒记为ACN。
1.2 Co-Cu/ACN催化剂的制备
1) 等体积浸渍法。取不同质量(0.49,1.47,2.45和4.43 g)的Co(NO3)2·6H2O溶于200 mL去离子水中,将配置好的溶液逐滴加到5.00 g ACN中,采用超声波辅助浸渍1 h,在110 ℃下干燥12 h,并于400 ℃和N2气氛下煅烧4 h,所得样品记为w%Co/ACN(w=2,6,10,14)。
称取2.45 g Co(NO3)2·6H2O 和相应质量(0.38,1.14 和1.9 g)的Cu(NO3)2·3H2O 溶于200 mL 去离子水中,将配置好的混合液逐滴加入到5.00 g ACN中,采用超声波辅助浸渍1 h,在110 ℃下干燥12 h,并于400 ℃在N2气氛下煅烧4 h,所得样品记为y%Cu-10%Co/ACN(y=2,6,10)。
2) 沉淀-沉积法。称取2.45 g Co(NO3)2·6H2O和0.38 g Cu(NO3)2·3H2O(Co 与Cu 物质的量比为5:1)溶于200 mL 去离子水中,在搅拌状态下将5.00 g ACN缓慢加入到混合溶液中,逐滴加入氨水溶液直至pH为10,继续搅拌3 h,在110 ℃下干燥12 h,并于400 ℃和N2气氛下煅烧4 h,所得样品记为Cu-Co/ACN-DP。
3)水热法。称取2.45 g Co(NO3)2·6H2O和0.38 g Cu(NO3)2·3H2O(Co与Cu物质的量比为5:1)溶于200 mL去离子水中,将溶液与5 g ACN同时加入到水热反应釜中,在120 ℃下处理12 h,在110 ℃下干燥12 h,并于400 ℃和N2气氛下煅烧4 h,所得样品记为Cu-Co/ACN-HY。
4) 溶胶-凝胶法。称取2.45 g Co(NO3)2·6H2O和0.38 g Cu(NO3)2·3H2O(Co 与Cu 物质的量比为5:1)溶于200 mL 去离子水中,在搅拌状态下将5.00 g 的ACN缓慢加入到混合溶液中,逐滴加入C6H8O7溶液后,在80 ℃的水浴条件下继续搅拌3 h,经110 ℃干燥12 h后,于400 ℃和N2气氛下煅烧4 h,所得样品记为Cu-Co/ACN-SG。
1.3 催化剂活性的评价
本实验采用模拟固定床反应器测试不同材料的烟气净化效果。图1所示为实验系统示意图。由图1可见:实验分为5个部分,即配气系统、控制系统、反应系统、采样测试系统和尾气处理系统。模拟烟气由配气和控制系统配置,模拟烟气经管路通入到反应系统中,在恒定温度下发生催化反应,反应后气体依次经过采样测试系统及尾气处理系统。模拟烟气体积空速为30 000 h-1,总流量为300 mL/min,O2体积分数为10%,CO质量浓度为6 g/m3,Hg0质量浓度为50µg/m3。
配气和控制系统包括气源和质量流量计,质量流量计精准控制气体流量,气源包括N2,O2和CO,其中,N2被分为2 个部分:一部分作为汞蒸汽发生系统的载气,另一部分作为整个气路的平衡气体。反应系统包含1 个内径为7 mm 的石英反应管和数显恒温炉,采样测试系统分为汞检测系统和CO检测系统。汞检测系统包括采样器、吸收液、还原剂和测汞仪。测定过程包括:气体中各种价态的汞(Hg0和Hg2+)在吸收液(KMnO4和H2SO4混合溶液)中转变为Hg2+;Hg2+在翻泡瓶中被还原剂(质量分数为15%酸性SnCl2溶液)转换为Hg0;Hg0在JKG-203型冷原子测汞仪中测定,经换算得到气体中Hg0浓度。CO 检测系统为Kane-9106 型烟气分析仪,对于CO 和O2的检测精度分别为1×10-6和1%。最后,反应结束后的尾气经炭罐净化后排放至室外。
催化材料的Hg0脱除率η(Hg0)通过下式计算:
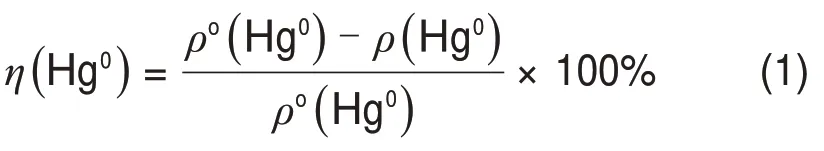
式中:ρo(Hg0)为反应系统进口Hg0质量浓度;ρ(Hg0)为反应系统出口Hg0质量浓度。
催化材料的CO脱除率η(CO)通过下式计算:
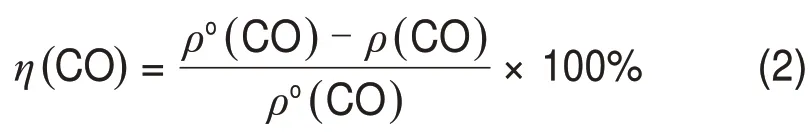
式中:ρo(CO)为反应系统进口CO 质量浓度;ρ(CO)为反应系统出口CO质量浓度。
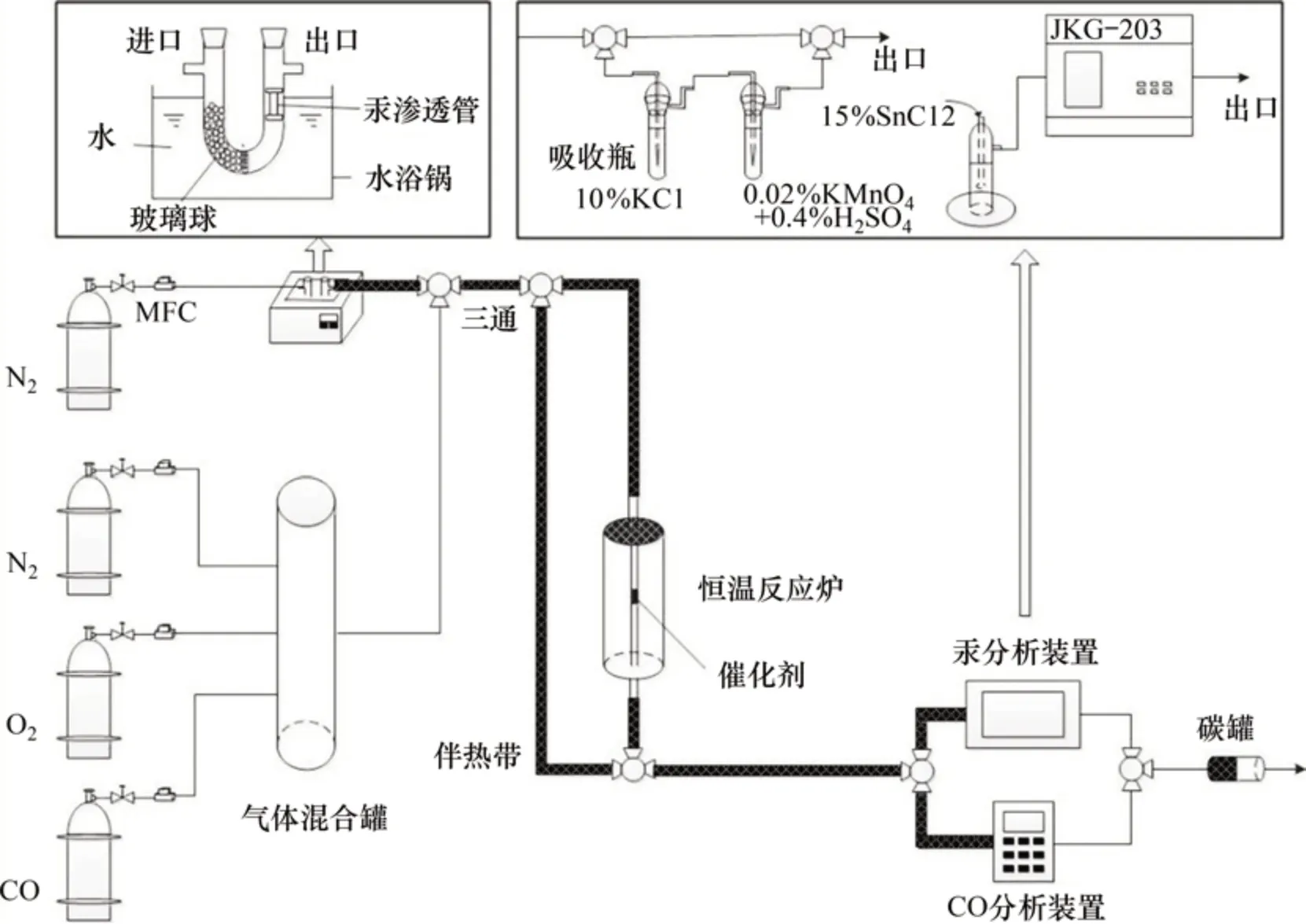
图1 实验系统示意图Fig.1 Schematic diagram of experimental system
1.4 催化材料的表征
1)通过物理吸附仪(Quadrasorb SI)测试可以得到催化剂比表面积、孔径分布孔容和孔道类型等信息,为进一步分析材料表面结构与性能的关系提供了更加详实的依据。
2)采用美国安捷伦7500ce 电感耦合等离子体质谱仪(ICP),根据处于激发态的待测元素原子回到基态时发射的特征谱线对催化剂中各元素进行定性及定量分析。
3)采用多功能X线衍射仪(TTRⅢ)测试催化剂XRD 谱图,通过对材料进行X 线衍射,大量原子散射波叠加、相互干涉而产生最大强度的光束,利用分析器衍射图谱,获得材料的成分、材料内部原子或分子结构等信息。
4)采用PCA-1200化学吸附分析测试仪进行测定。实验前准确称取0.1 g 待测样品放入石英管反应器中,在流量为100 mL/min的O2/He吹扫气氛条件下高温预处理1 h后,冷却至室温,采用100 mL/min的H2/Ar吹扫至基线稳定后,以10 ℃/min的升温速率升至目标温度,根据得到的谱图分析催化剂氧化还原性能。
5) 采用傅里叶变换红外光谱仪(Thermo Nicolet IS50),利用干涉光通过样品池中含有材料信息的干涉光到达检测器;然后,通过傅里叶变换对信号进行处理;最终,根据得到的光谱图分析其表面官能团。
2 实验结果
2.1 Co负载量对催化剂性能的影响
采用等体积浸渍法制备了一系列不同钴负载量的活性焦催化剂,其Hg0和CO 脱除率测试结果如图2所示。从图2可见:随着钴负载量增加,改性活性焦的CO 脱除率得到有效提升,而Hg0脱除率提升较小。适当的负载量(10%)可以保证活性组分在催化材料表面的均匀分散,这是实现高效催化的重要条件之一,而随着负载量进一步增加,活性组分发生堆积,分散性被破坏,抑制了催化反应,最终导致脱除率下降。由实验结果可以推测:Co 改性的活性焦材料可以大量提供CO 与O2的反应活性位点,而不会或很少提供Hg0反应活性位点。
2.2 添加Cu对催化剂性能的影响
负载Co 后活性焦催化剂的CO 脱除率大幅提升,但Hg0脱除率仍然未取得较大的提升。XU等[19]人制备了一系列CuO/TiO2催化剂,考察催化剂在模拟烟气条件下的Hg0氧化性能,发现Cu2+可以有效促进Hg0的催化氧化,因此,进一步向10%Co/ACN催化剂中引入不同质量分数的Cu,以提高双功能催化剂的催化效率,效率测试结果如图3所示。由图3可见:加入2%的Cu组分后,活性焦催化剂的Hg0脱除率显著提高,而CO 脱除率提升较小。当Cu 组分大于2%时,随着质量分数进一步增加,催化效率下降。由此推测:Cu 适度添加为活性焦材料引入了Hg0和CO 的反应活性点位,尤其是增加了大量脱Hg0反应活性位,但Cu 组分的引入会导致活性焦表面的活性点位分布发生变化,最终致使Hg0脱除率和CO 脱除率下降。当反应温度为140~200 ℃时,10%Co~2%Cu/ACN获得最佳的同时Hg0脱除率和CO 的脱除率。在200 ℃时,10% Co~2% Cu/ACN可以达到80.0%的Hg0脱除率以及99.7%的CO脱除率,比10%Co/ACN的Hg0脱除率提高了近20%。
2.3 不同制备方法对催化剂性能的影响
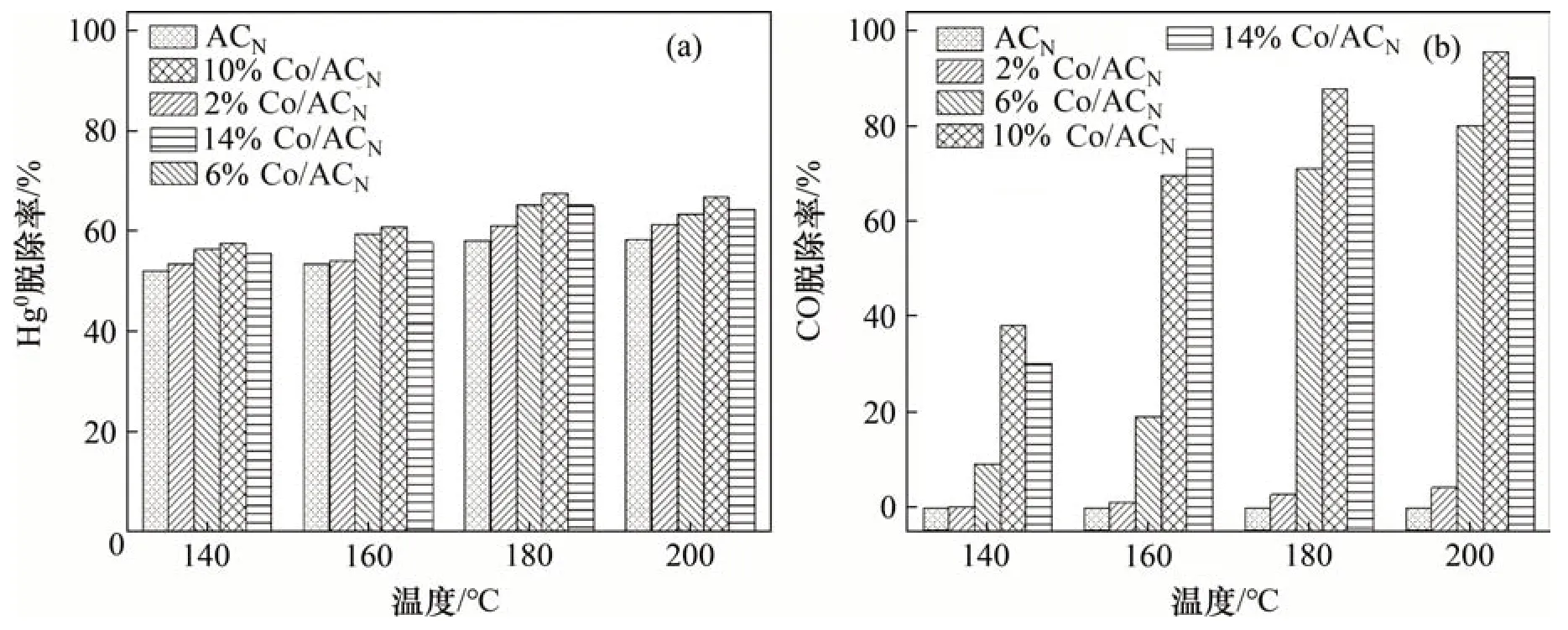
图2 不同Co负载量的活性焦催化剂活性测试结果Fig.2 Activity test results of activated coke catalysts with different Co loadings
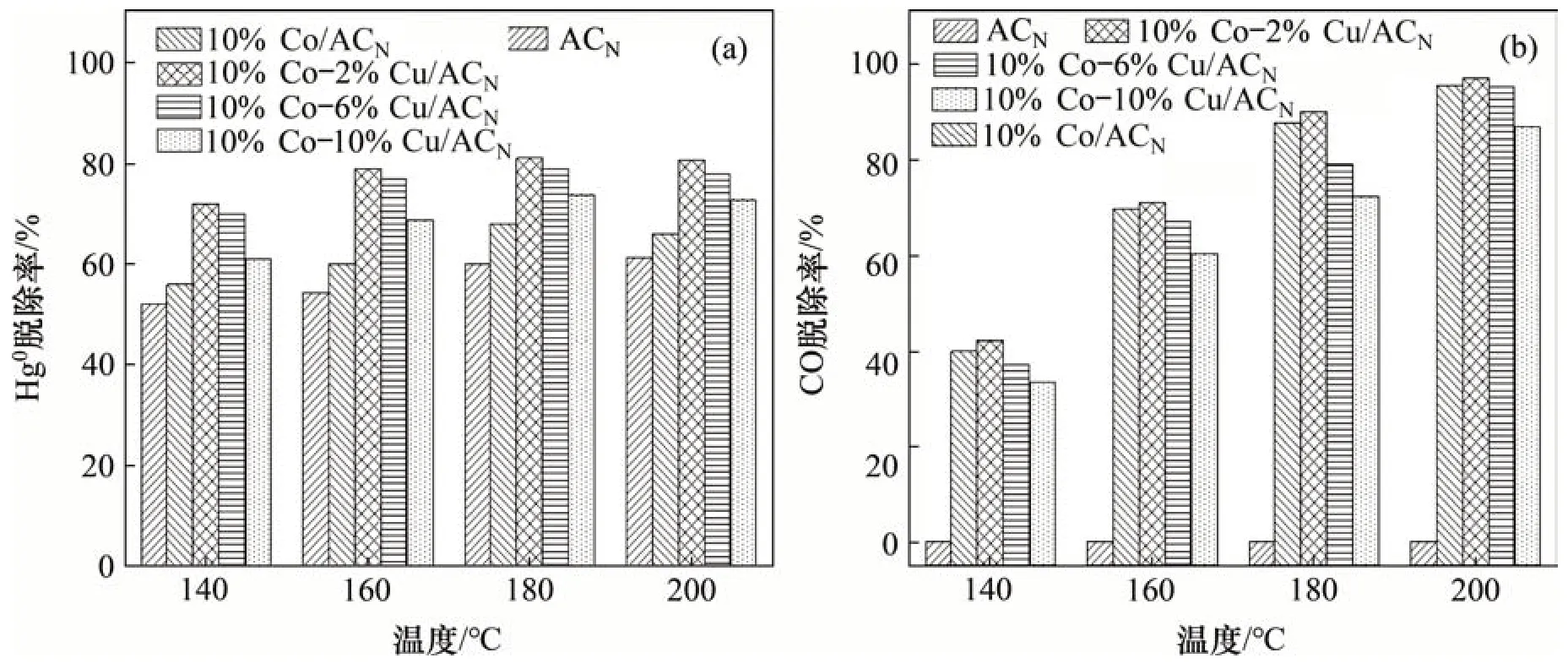
图3 掺杂铜元素后催化剂的活性测试结果Fig.3 Activity test results of Cu-doped catalysts
活性组分负载量以及负载方式都是影响活性焦催化剂催化效率的关键因素,不同的制备方式下产生氧化物的种类以及分散程度不同。图4所示为4 种方法制备的2% Cu~10% Co/ACNHg0脱除率和CO 脱除率(在图中简称为Cu-Co/ACN)。由图4(a)可知:在140~160 ℃时,4 种催化剂的Hg0脱除率由高到低依次为Cu-Co/ACN-SG,Cu-Co/ACNHY,Cu-Co/ACN-VI 和Cu-Co/ACN-DP;在160~200 ℃时,Cu-Co/ACN-SG和Cu-Co/ACN-HY催化剂的Hg0脱除率高于其他2 种催化剂,而Cu-Co/ACN-DP 与Cu-Co/ACN-VI 的Hg0脱除率相近。在测试温度区间内,Cu-Co/ACN-SG 可以取得大于90%的Hg0脱除率,最高效率可达96%(180 ℃);Cu-Co/ACN-HY 可以获得85%左右的Hg0脱除率,且随着反应温度提升,效率几乎保持不变;Cu-Co/ACN-DP 和Cu-Co/ACN-VI 可以获得相近的Hg0脱除率(70%~80%)。
由图4(b)可见:4 种催化剂的脱CO 效率均随着反应温度上升而提高,在180~200 ℃时,其CO脱除率从高到低依次为Cu-Co/ACN-VI,Cu-Co/ACN-DP,Cu-Co/ACN-SG 和Cu-Co/ACN-HY。其中,Cu-Co/ACN-HY 在实验温度范围内几乎不具备脱CO 能力;Cu-Co/ACN-DP 和Cu-Co/ACN-SG仅可以获得小于40%的CO 脱除率,活性温窗窄;Cu-Co/ACN-VICO 脱除率最佳,当反应温度为200 ℃时,CO脱除率可达99%。
综上所述:Cu-Co/ACN-VI 催化剂同时脱Hg0和CO 性能最佳,在160~200 ℃内可以取得大于70%的Hg0脱除率和大于70%的CO 脱除率,适用于烧结烟气同时脱Hg0脱CO 需求。当反应温度从180 ℃增加到200 ℃时,催化剂的CO 脱除率提升25%,Hg0脱除率仅仅下降2%,因此,在200 ℃的反应条件下,等体积浸渍法制备的Cu-Co/ACN催化剂取得最佳同时Hg0脱除率(76%)和CO 脱除率(99%)。
2.4 氧体积分数对催化剂的影响
在实际钢铁工业中,烧结烟气的氧体积分数波动性大,为13%~20%。因此,在200 ℃的反应条件下,进一步考察氧体积分数(0%,4%,8%,12%,16%和20%)对Cu-Co/ACN-VI催化剂同时脱Hg0和脱CO的影响,测试结果如图5所示。由图5可见:无氧条件下,对Hg0和CO 的脱除率很小;当氧体积分数提升到4%时,催化剂对Hg0和CO的脱除率分别提升到51%和91%,这表明氧气对催化反应具有显著影响;当氧体积分数继续提升至16%时,催化剂对Hg0和CO 的脱除率达到最大,随后保持稳定。当O2体积分数为16%时,催化剂对Hg0的脱除率(77.6%)和对CO的脱除率(99.9%)最高。一般认为,O2能够补充催化剂表面被消耗的晶格氧或化学吸附氧,有利于吸附态Hg0生成氧化态汞[20],同时为CO 催化氧化提供活性氧物种[21],因此,提高了催化剂同时脱Hg0和脱CO 性能。综上所述:该催化剂适用于13%~20%氧体积分数的烧结烟气净化。
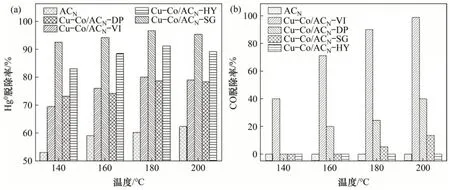
图4 催化材料活性测试结果Fig.4 Catalytic material activity test results
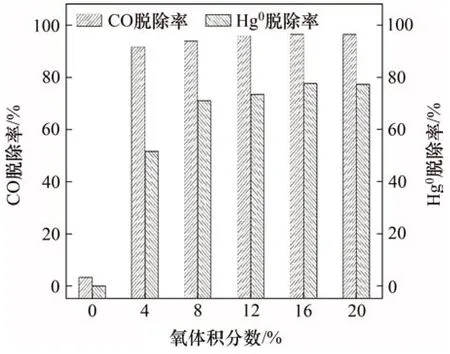
图5 200 ℃条件下氧体积分数对同时脱Hg0脱CO性能的影响Fig.5 Effect of oxygen volume fraction on CO and Hg0 removal performance with reaction temperature of 200℃
3 讨论与分析
3.1 催化剂孔隙结构
表1所示为不同方法制备的Cu-Co/ACN催化剂与载体ACN孔隙结构参数的对比,由表1可见:总孔比表面积由高到低依次为ACN,Cu-Co/ACNSG,Cu-Co/ACN-HY,Cu-Co/ACN-VI 和Cu-Co/ACN-DP;而微孔比表面积由高到低依次为Cu-Co/ACN-VI,Cu-Co/ACN-DP,Cu-Co/ACN-SG,Cu-Co/ACN-HY 和ACN。这表明在活性组分在负载过程中,虽然活性焦原有的孔道结构部分堵塞,总比表面积下降,但同时也产生了部分微孔结构。相比中孔而言,微孔更有利于烟气中对Hg0吸附[22]。
3.2 催化剂物相结构
图6所示为4种方法制备的Cu-Co/ACN催化剂的XRD 图谱。4 种催化剂分别存在CuO[6],Co3O4[23],Co2O3和CoO[23]特征峰,不存在金属硝酸盐的特征峰,这表明硝酸金属盐在相应的焙烧温度下全部转化为金属氧化物。
由图6可知:Cu-Co/ACN-VI表面的主要组分为钴氧化物,未见到明显的铜氧化物的衍射峰,推测该制备方法下铜氧化物在载体表面分散较均匀;不具备脱CO能力的Cu-Co/ACN-HY表面主要钴氧化物为CoO,CO脱除率较高的Cu-Co/ACN-VI表面主要钴氧化物为Co3O4和Co2O3,由此推测在钴氧化物中,Co3O4和Co2O3为CO 的脱除提供了主要的活性位点。由于CuO对Hg0具有极强的吸附作用[24],吸附态汞增多有利于Hg0的催化氧化,而Hg0脱除率较高的Cu-Co/ACN-SG 与Cu-Co/ACN-HY 的主要铜氧化物为CuO,因此,本研究中CuO为Hg0脱除提供了主要的活性位点。
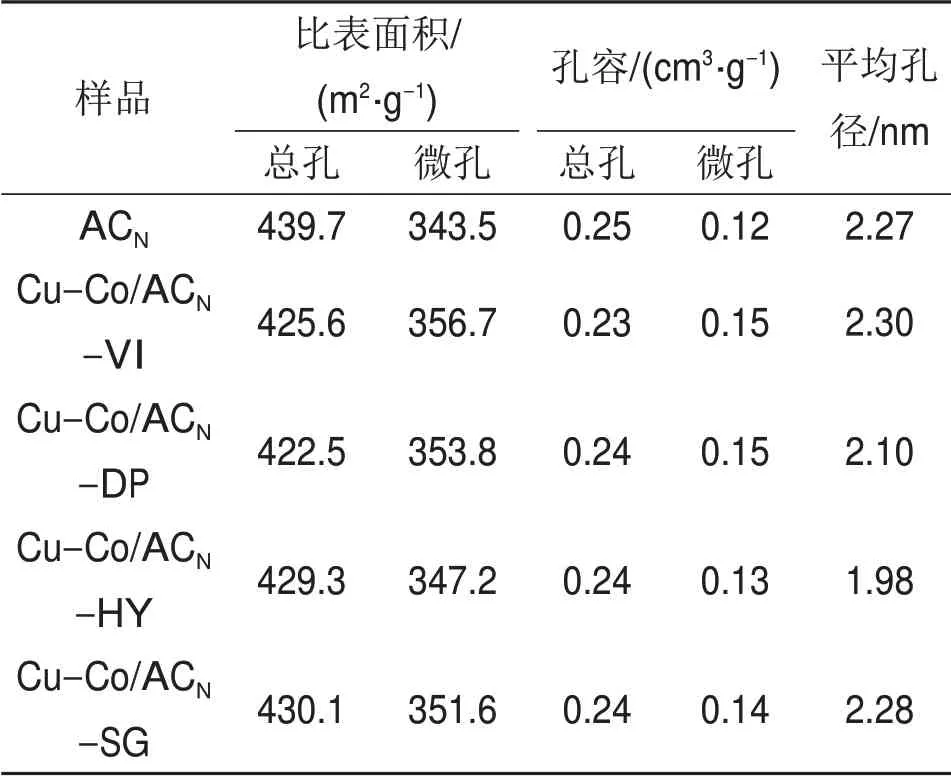
表1 催化剂BET结果Table 1 BET results of catalysts
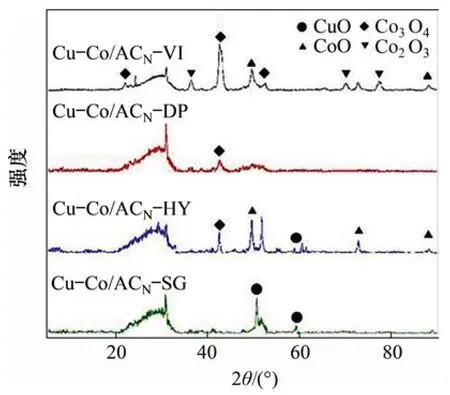
图6 催化剂XRD谱图Fig.6 XRD pattern of catalysts
3.3 催化剂氧化还原能力
4种样品的H2-TPR谱图如图7所示。从图7可见:Cu-Co/ACN-VI和Cu-Co/ACN-DP催化剂在还原过程中经历了3 个不同阶段:Co2O3→Co3O4,Co3O4→CoO 和CoO→Co[25],分别对应240,334 和456 ℃附近的3个峰。Cu-Co/ACN-HY催化剂则具备2 个还原峰,分别对应CuO→Cu(358 ℃)和CoO→Co(484 ℃)还原转化;Cu-Co/ACN-SG 催化剂仅具备1个还原峰(367 ℃),对应CuO→Cu还原转化[26]。因此,Cu-Co/ACN-VI和Cu-Co/ACN-DP催化剂表面含有丰富的钴氧化物,缺乏铜氧化物,与XRD观察到的结果一致;Cu-Co/ACN-HY催化剂同时拥有钴氧化物和铜氧化物;Cu-Co/ACN-SG催化剂中铜氧化物占主导位置,而钴氧化物可能由于负载量较小或分散较均匀,未被观察到。
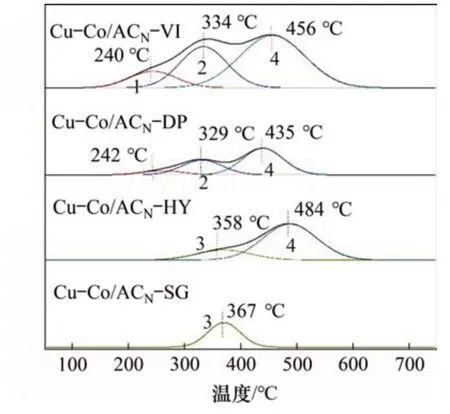
图7 催化剂的氢气程序升温还原(H2-TPR)谱图Fig.7 Hydrogen temperature-programmed reduction(H2-TPR)profiles of catalysts
4 种样品的H2-TPR 分析结果如表2所示,钴氧化物还原峰分别标记为1 号、2 号和4 号,铜氧化物还原峰标记为3号。Co2O3→Co3O4过程中部分Co3+被还原为Co2+,Co3O4→CoO过程中剩余Co3+被还原为Co2+,CoO→Co 过程中全部Co2+被还原为Co。由表2可知:Cu-Co/ACN-VI 催化剂的4 号还原峰面积是1 号和2 号还原峰面积之和的1.98 倍,而Cu-Co/ACN-DP 催化剂的还原峰面积为1 号和2号峰面积之和的2.03倍。Cu-Co/ACN-VI催化剂的2号还原峰面积是1号还原峰面积的2.94倍,而Cu-Co/ACN-DP 催化剂中的还原峰面积为1 号还原峰面积的3.31倍,这表明2个催化剂表面的主要钴氧化物为Co3O4。Cu-Co/ACN-VI催化剂表面Co3O4的还原峰面积是Cu-Co/ACN-DP 的3.06 倍,这导致Cu-Co/ACN-VI 催化剂CO 脱除率分别为Cu-Co/ACN-DP 的3.21 倍(160 ℃)和3.05 倍(180 ℃)。Cu-Co/ACN-HY催化剂在还原过程中存在CoO→Co还原转化过程,表面的钴氧化物主要为CoO,而该物质不是脱CO 过程的活性位点。3 号峰表示催化剂中CuO→Cu 的还原转化,Cu-Co/ACN-SG 催化剂表面的CuO 还原峰面积是Cu-Co/ACN-HY 还原峰面积的1.41倍,而Cu-Co/ACN-SG催化剂Hg0脱除率是Cu-Co/ACN-HYHg0脱除率的1.06~1.08 倍。综上所述:Co3O4和CuO分别是脱CO和脱Hg0过程的主要活性位点。
3.4 催化剂表面观能团
为进一步分析活性焦催化材料表面官能团的差异,分别采取了4种样品的FTIR图谱,如图8所示。通常来说,不同位置的峰值对应不同的官能团:3 420 cm-1处的峰值对应于材料表面吸附水的羟基O—H 振动;2 929 cm-1和2 851 cm-1处的峰值分别对应C—H 对称和不对称振动;1 578 cm-1处的峰值对应芳香环中的C—C振动;1 128 cm-1处的峰值对应羧基O—C=O振动;1 022 cm-1处的峰值对应C—O振动[27];672 cm-1和565 cm-1处的峰值则对应Co—O振动[28]。通过比较发现:4种材料表面官能团的差异主要集中于含氧官能团(O—C=O及C—O)和Co—O 官能团。由图8可见:CO 脱除率最高的Cu-Co/ACN-VI 显示出最强的Co—O 振动峰。XRD结果中,Cu-Co/ACN-VI同样表现出最强的钴氧化物衍射峰以及最多的钴氧化物种类,由此表明钴氧化物有利于CO催化氧化反应。含氧官能团在图8中差异不明显,可能由于活性焦表面本身具有较为丰富的官能团类型,因此,由制备方法所带来的含氧官能团变化相比原有焦炭表面强度较弱。
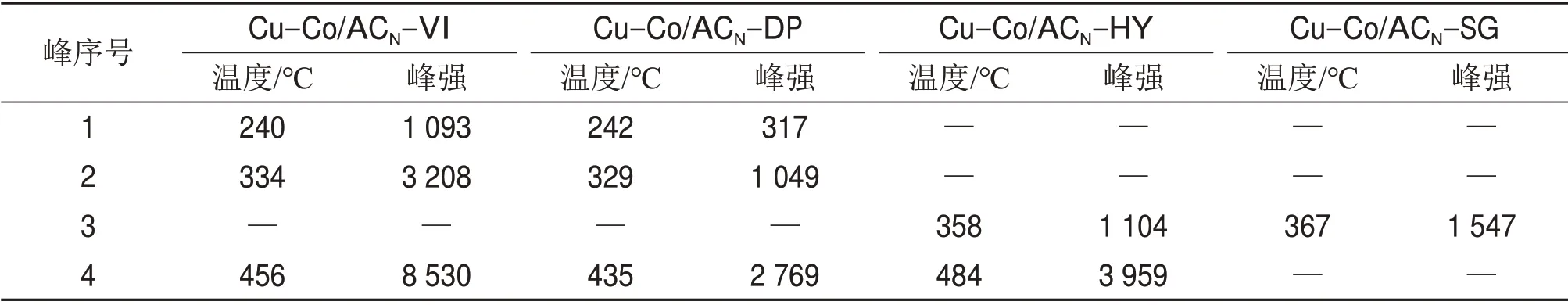
表2 H2-TPR分析结果Table 2 Analysis results of H2-TPR
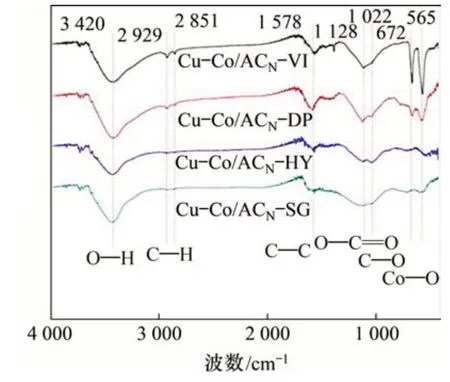
图8 催化剂的傅里叶红外变换(FTIR)光谱图Fig.8 Fourier-transform infrared(FTIR)spectra of catalysts
4 结论
1)硝酸改性活性焦具备脱Hg0能力,但不具备脱CO 能力。Co 和Cu 的添加有利于增加催化剂表面活性位点数量,提高了催化剂同时脱Hg0脱CO性能。
2)在200 ℃下,采用等体积浸渍法制备的2%Cu-10%Co/ACN催化剂同时脱Hg0和CO 的效果最佳,且催化剂适用于氧气波动较大的条件。
3)与沉淀-沉积法、水热合成法和溶胶-凝胶法相比,等体积浸渍法制备的催化剂表面存在更多的Co3O4和CuO活性物种,提升了催化剂的氧化还原能力,从而促进催化反应,最终具有最佳同时脱Hg0脱CO性能。
猜你喜欢
化工管理(2022年13期)2022-12-02
消费电子(2022年6期)2022-08-25
石油化工高等学校学报(2022年1期)2022-04-15
陶瓷学报(2021年4期)2021-10-14
建材发展导向(2021年12期)2021-07-22
烟台果树(2021年3期)2021-07-21
陶瓷学报(2021年1期)2021-04-13
陶瓷学报(2020年6期)2021-01-26
船舶标准化工程师(2020年1期)2020-06-12
中外葡萄与葡萄酒(2019年2期)2019-04-12
