
基于叶绿体DNA变异的山荆子种质遗传多样性和系统演化
2020-02-25 08:25高源王大江王昆丛佩华张彩霞李连文朴继成
中国农业科学 2020年3期
高源,王大江,王昆,丛佩华,张彩霞,李连文,朴继成
基于叶绿体DNA变异的山荆子种质遗传多样性和系统演化
高源,王大江,王昆,丛佩华,张彩霞,李连文,朴继成
(中国农业科学院果树研究所/农业部园艺作物种质资源利用重点实验室,辽宁兴城 125100)
【目的】山荆子是中国原产苹果属植物中分布最广泛的种,母系遗传的叶绿体基因组的非编码区适用于较低的分类阶元(如科、属)的系统研究。对野外考察新收集的山荆子种质的叶绿体DNA(cpDNA)非编码区进行测序,解析其序列遗传变异,从母系遗传基因的角度探究山荆子不同居群的遗传多样性和系统演化关系,为我国山荆子种质资源的起源演化以及收集和保护提供理论依据。【方法】利用4对叶绿体DNA引物扩增新收集的215份山荆子种质资源的4个非编码区H-A、S-G spacer+intron、T-5'L和5'L-F,对每个基因间区正反向测序获得的序列进行人工校对后,使用MEGA 7.0进行序列拼接和比对,并构建山荆子不同居群间基于遗传距离的Neighbour-Joining系统发育树;使用DnaSP ver5.10.01计算叶绿体DNA的遗传多样性参数,计算不同居群间的基因流和基因分化;利用Arlequin v3.5分析标准分子变异(AMOVA);运用NetWork ver4.6.1.2构建种内居群间的叶绿体DNA单倍型邻接网络关联图。【结果】4个叶绿体DNA非编码区经测序、拼接、比对和合并之后的片段长度为3 777 bp,共有171个多态性变异位点,其中包含150个插入-缺失位点、20个简约信息位点和1个单一突变位点。在215份山荆子种质中,H-A、S-G spacer + intron、T-5'L和5'L-F区域的变异位点数量分别为26、32、103和10个,单倍型数量分别为8、8、6和4个,合并之后的叶绿体DNA片段的单倍型为24个。核苷酸多样性最高的区域为T-5'L(Pi=0.01174),单倍型(基因)多样性最高的为S-G spacer+intron(d=0.599),最低的为5'L-F(d=0.228)。215份山荆子种质叶绿体DNA多样性较高(d=0.727,Pi=0.00577)。Tajima’s D检验中,4个cpDNA区域在各检验水平上均不显著,检测的4个cpDNA区域在进化上遵循中性模型。AMOVA分析表明遗传变异主要存在于群体内部。【结论】供试4个叶绿体DNA非编码区适合苹果属山荆子种质遗传多样性和系统演化分析。在叶绿体DNA水平导致山荆子群体进化的原因不是自然选择,而是突变压力和遗传漂变。群体间遗传分化与其地理距离不完全相关。山荆子可能为多点起源,推测黑龙江和吉林,内蒙古,甘肃和山西为3个可能的起源地区。
山荆子;叶绿体DNA;非编码区;遗传多样性;系统演化
0 引言
【研究意义】山荆子(( L.) Borkh.),又称山定子[1],属蔷薇科(Rosaceae)苹果属(Mill.)[2]山荆子组(Sect.Koehne Ser.)山荆子系(Ser.Rehd.)[3]。山荆子在苹果属植物中分布最广泛,属东亚分布型,但多分布于我国境内[4],在近些年的野外考察地点都可以见到[5]。山荆子用途极广,在园林造景上广泛应用,在苹果生产上多用作砧木,还可用于抗性砧木的选育,是苹果属植物中经济价值较高的珍贵野生种。从多角度充分研究山荆子的遗传多样性和遗传基础是对丰富的山荆子野生资源开发利用的前提。基于野外考察收集到的苹果属植物,从母系遗传基因的角度研究苹果属植物的遗传多样性水平,探究其原生境的系统演化途径,对指导苹果属植物的收集和保护,探讨苹果属不同种的系统演化具有重要意义。【前人研究进展】山荆子原产于亚洲东北部,在长期演变和进化过程中形成了丰富的变异类型[6],现主要分布于中国的东北、华北及西南地区[7-9],其现代高度适生区为山西太行山、管涔山和吕梁山,吉林和辽宁东北部,陕甘宁交界处,河北北部和鲁中南地区[10]。山荆子表型多样性变异丰富,居群遗传分化和亲缘关系复杂[11-16]。从分子角度,以往利用RAPD[13]、ISSR[17]和SSR[18-19]分子标记方法对多个山荆子居群的遗传多样性进行研究,推测山荆子起源于中国华北和东北[18],河北山荆子的遗传多样性水平最高,并且主要在群体内和个体内部发生遗传变异和分化[19]。以往主要利用表型和少量基因组分子标记研究其遗传多样性,尚未从母系遗传基因的角度对山荆子群体之间的遗传多样性和亲缘关系进行解析。邹旭等[10]从气候变化的角度对末次盛冰期以来的中国山荆子分布格局进行了预测,但是现存的山荆子各居群间的系统演化关系则需要更多基因变异方面的证据加以验证。叶绿体基因组高度保守,非编码区进化速度较快,适用于较低的分类阶元(如科、属)的系统研究[20]。在植物中利用叶绿体基因组序列可直接进行遗传多样性检测[21],在植物系统发育研究中,叶绿体基因组母系遗传基因的特点具有独特优势[22],在植物系统发育、遗传多样性和亲缘演化关系等研究方面发挥着重要作用[23]。国外学者利用一至两个叶绿体DNA(cpDNA)区域较早地开展了苹果属栽培品种和野生近缘种的叶绿体DNA变异和亲缘关系等的研究[24-27],丁芳兵[28]利用叶绿体基因k与核糖体基因相结合研究湖北海棠与近缘种的亲缘关系;朱元娣等[29]利用相同的基因区域研究了新疆野苹果与中国苹果的系统发育,并得出的叶绿体基因k并不适用于栽培苹果种内系统发育分析的结论;徐榕雪[30]也利用该区域的内含子序列信息,构建了苹果属23个种共27份材料系统发育树,但仅获得遗传变异信息位点23个,推断k不太适合近缘种间及种下类群材料的遗传关系分析。李慧峰[31]利用L-F序列构建的泰沂山区苹果属植物系统树在一定程度上可以支持表型分类结果,但无法有效解决其复杂遗传背景下的系统发育关系。叶绿体单个基因或单个基因间隔区难以满足具有复杂遗传背景的果树种质资源的遗传分析要求,叶绿体基因组用于系统发育的研究已经发展到多个基因及基因间隔区多态性位点组合分析的阶段[32-34]。由此可见,要利用叶绿体DNA对具有复杂遗传背景的苹果属植物的遗传变异分析,需要用大群体、进化速率快的多个区域才可实现。【本研究切入点】随着叶绿体DNA研究技术的应用和发展,基于2008—2015年中国农业科学院果树研究所国家果树种质兴城梨、苹果圃野外考察新收集入圃的苹果属山荆子种质资源,在前期利用表型鉴定其代表性和SSR分子标记遗传评价的基础上[19],研究其叶绿体DNA多个非编码区的遗传变异,从母系遗传基因的角度进一步揭示山荆子的遗传多样性和系统演化。【拟解决的关键问题】本研究对新收集的215份山荆子种质资源的4个非编码区利用4对通用的叶绿体DNA引物进行扩增,分析4个叶绿体DNA间区序列的遗传变异,解析山荆子群体在叶绿体DNA水平上的遗传多样性,探讨7个山荆子居群间的系统演化关系,为山荆子的起源演化及收集和保护提供依据。
1 材料与方法
1.1 材料
供试种质中MB84—MB101采自国家果树种质公主岭寒地果树圃(吉林省公主岭),其余种质均采自国家果树种质兴城梨、苹果圃(辽宁省兴城市)。2008—2015年,多次赴山荆子的主要分布区域内蒙古、黑龙江、吉林、辽宁、河北、山西和甘肃等地进行考察和收集,在野外通过表型鉴定为山荆子本种后,记录种质地理信息,采集表型差异较大的种质接穗,分别入两个圃嫁接繁殖,育成植株作为种质保存。于2017年春季采集山荆子种质的健康幼嫩叶片,叶片经硅胶干燥之后备用;2018年完成所有材料4个叶绿体DNA非编码区测序,所有供试山荆子材料信息见电子附表1。215份材料包括来自于内蒙古42份、黑龙江83份、河北54份、吉林18份、山西13份、甘肃4份和辽宁1份。
1.2 DNA提取
采用德国QIAGEN的DNeasy Plant Mini Kit试剂盒提取供试材料春季嫩叶的基因组DNA。分别用1%的琼脂糖凝胶电泳和紫外分光光度计检测其浓度和纯度,经检验合格后,一部分提取DNA原液-20℃冷冻保存,一部分原液稀释至40 ng·μL-1备用。
1.3 PCR扩增
为保证双向测序能够测通叶绿体通用引物的扩增产物,从Shaw等[35]报道的叶绿体DNA序列中选取无回文结构的基因区间和扩增片段长度在1 500 bp以下的叶绿体通用引物,4个基因间区H-A、S-G spacer + intron、T-5'L和5'L-F对应的4对叶绿体通用引物(表1)由上海生工(Sangon)有限公司合成。50 μL的PCR反应体系:DNA模板2 μL,正向和反向引物(10 µmol∙L-1)各2.5 μL,d NTP(2.5 mmol∙L-1)(Takara,Japan)5 μL,10×Buffer 5 μL,Taq DNA聚合酶(5 U∙μL-1)(Takara,Japan)0.6 μL,ddH2O 32.4 μL。参照Volk等[36]报道的反应条件,对4个基因间区的PCR扩增条件进行优化(表2)。215份种质4个叶绿体间区的PCR产物用2%的琼脂糖凝胶电泳检测(图1),对于扩增成功并得到单一条带的样品直接送测序公司(上海美吉测序公司(北京))进行测序,测序仪为美国ABI 3730 DNA Sequencer。每个样品在每个叶绿体DNA间区的扩增产物均进行正、反向测序,215份材料的4个间区的叶绿体DNA序列全部测通。
1.4 数据分析
每个基因间区正、反向测序获得的序列进行人工校对,并使用MEGA 7.0[37]软件进行序列拼接、序列比对以及4个叶绿体DNA基因间区片段H-A、S-G spacer+intron、T-5'L和5'L-F的合并。所有拼接序列、比对序列都被保存为FASTA和MEGA格式。

表1 4个叶绿体基因间区及4对叶绿体DNA引物序列
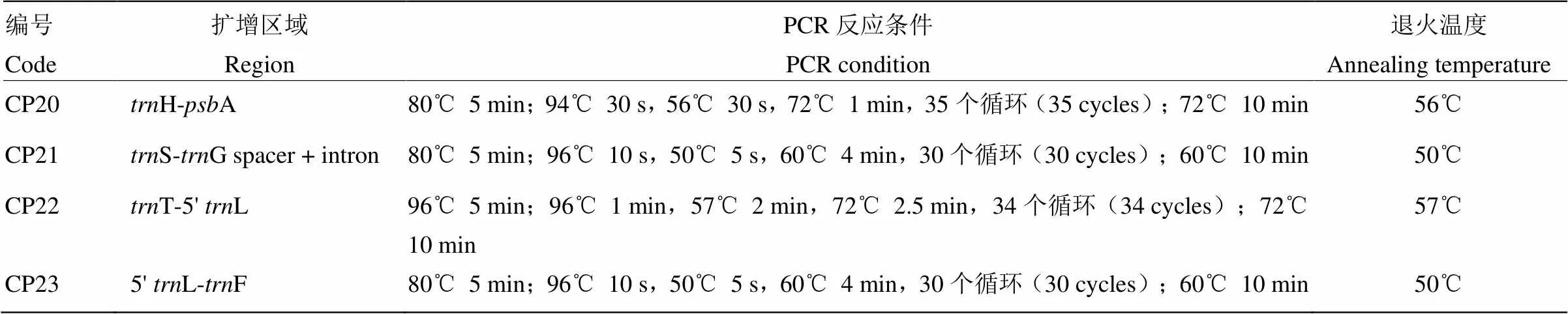
表2 山荆子4个叶绿体基因间区的PCR扩增
图1 部分苹果样品在叶绿体DNA间区trnH-psbA的PCR扩增产物的2%琼脂糖凝胶电泳图
将单个区域片段和合并之后的片段保存后,使用DnaSP ver5.10.01[38]软件计算山荆子种质叶绿体DNA的遗传多样性参数,包括:变异位点(Vs)、单一突变位点(Ss)、简约信息性位点(Ps)、核苷酸多样性(Pi)、平均核苷酸差异(k)、单倍型数目()、单倍型多样性(d)、单倍型多样性方差(Vh)、单倍型多样性标准差(Sh)和Tajima’s D值(Tajima’s D)。Tajima’s D值是检验样品叶绿体DNA区域是否遵循中性进化模型的重要参数。基于4个叶绿体DNA区域合并片段的山荆子种质资源的叶绿体基因单倍型保存为Nexus文件,分别利用中介邻接网络(median-Joining network,MJ)算法和最大简约(maximum parsimony calculation,MP)算法进行计算和优化,运用NetWork ver4.6.1.2构建种内居群间的叶绿体DNA单倍型邻接网络关联图。
使用MEGA 7.0软件构建山荆子种内居群间基于遗传距离的Neighbour-Joining(NJ)[39]系统发育树,并划分居群组合。使用DnaSP ver5.10.01软件计算种内不同居群间的基因流和基因分化,评价种内不同居群间的基因交流。使用Arlequin v3.5[40]软件分析分子变异,选择“标准分子变异分析”(standard molecular variation analysis,AMOVA)方法进行计算,用20 000次排列来检测不同分组变异的差异显著性,评价种群内居群间的遗传分化。
2 结果
2.1 山荆子叶绿体DNA的多态性
4对叶绿体通用引物对215份中国原产苹果属植物山荆子的4个叶绿体DNA间区进行扩增,H-A、S-G spacer + intron、T-5'L和5'L-F在所有样本中的序列长度范围分别为261—278 bp、1 367—1 383 bp、1 064—1 093 bp和957—965 bp,进行序列比对后,4个区域的片段长度分别为285、1 383、1 140和965 bp。其中,区域T-5'L核苷酸多态性最高,有92个插入-缺失位点和11个简约信息性位点;285 bp的非编码区H-A有2个简约信息位点和24个插入-缺失位点,区域spacerintron有1个单一突变位点、5个简约信息位点和26个插入-缺失位点;区域5'L-F的核苷酸多态性最低,简约信息性位点和插入-缺失位点分别仅有2个和8个。4个叶绿体基因间区序列组合之后长度范围为3 675—3 693 bp,经序列比对之后的长度为3 773 bp,含有150个插入-缺失位点、20个简约信息性位点和1个单一突变位点,共计171个多态性变异位点(表3)。
215份中国原产苹果属植物山荆子的4个叶绿体间隔间区共有变异位点171个,区域H-A、S-G spacer+intron、T-5'L和5'L-F的变异位点分别为26、32、103和10个,区域T-5'L含有其中60.2%的变异位点,是山荆子4个叶绿体DNA间区中多态性较高的区域。核苷酸多样性(Pi)和平均核苷酸差异(k)最大和最小的区域分别为T-5'L(Pi=0.01174,k=15.68937)和5'L-F(Pi=0.00045,k=0.44216)。H-A、S-G spacer+intron、T-5'L和5'L-F区域单倍型数目()分别为8、8、6和4个,以S-G spacer+intron区域单倍型(基因)多样性(d)最高为0.599,但其5'L-F单倍型(基因)多样性(d)最低为0.228。区域T-5'L的单倍型多样性方差和单倍型多样性标准差分别为0.00168和0.041,均为4个区域中最高值。区域S-G spacer+intron单倍型多样性虽然最高,但其单倍型多样性方差和标准差均为最低,分别为0.00111和0.033(表4)。
215份山荆子的4个叶绿体DNA间区组合之后的片段共有24个单倍型,各种质所属单倍型见电子附表1。核苷酸多样性、平均核苷酸差异、单倍型多样性、单倍型多样性方差和标准差分别为0.00577、23.79039、0.727、0.00084和0.029。
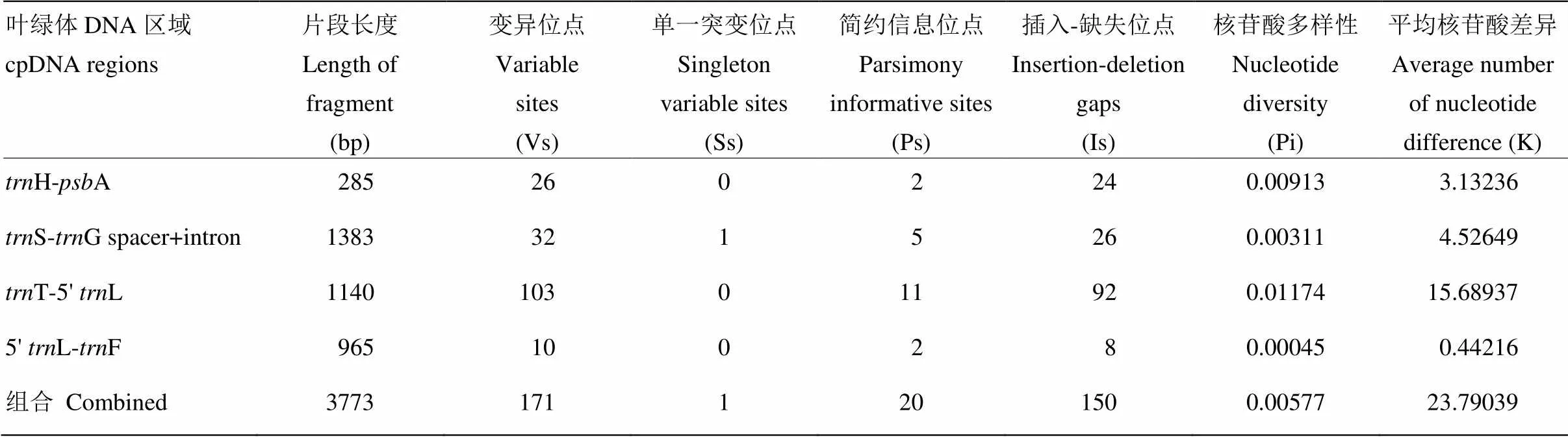
表3 供试山荆子的4个cpDNA区域的多态性信息
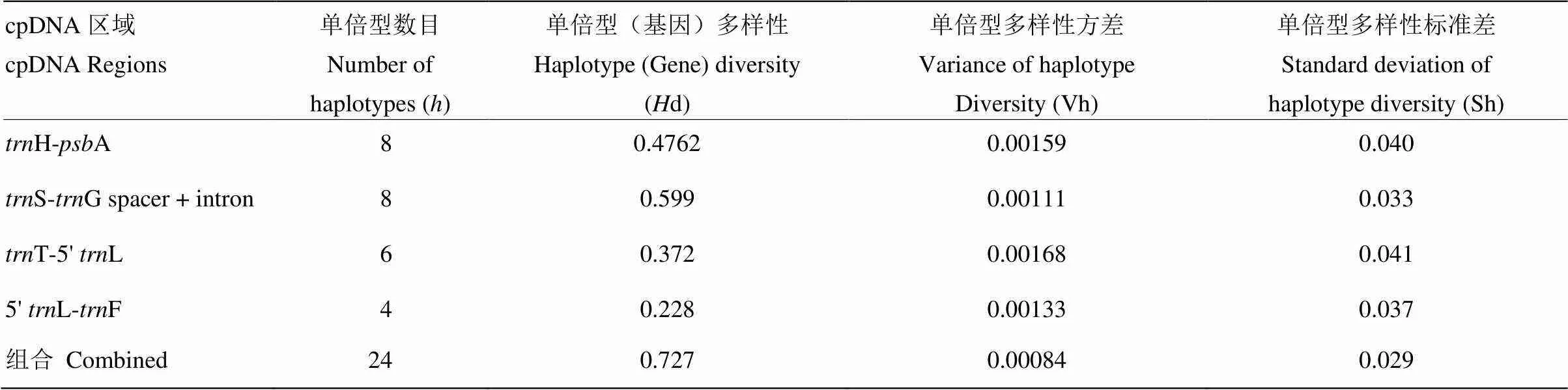
表4 供试山荆子叶绿体DNA单倍型多样性
对4个区域进行Tajima’s测验,Tajima’s D值全部为负值,区域5'L-F的Tajima’s D值最低,为-1.70516,区域T-5'L的Tajima’s D值最高,为-0.29083。除区域5'L-F的D值在0.05<<0.10>水平上不显著,其余3个区域的D值均在>0.10水平上不显著。4个区域合并之后,Tajima’s D值为-0.54177,在>0.10水平上不显著(表5)。
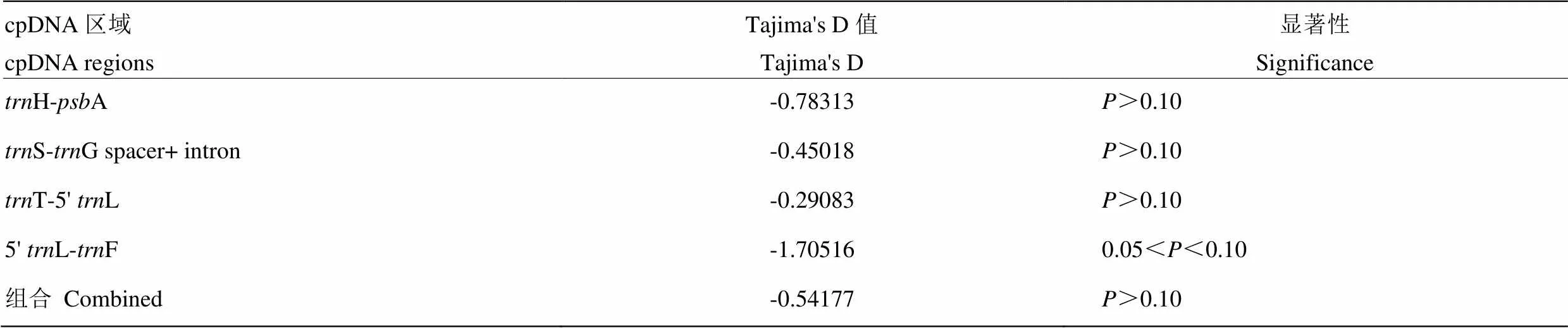
表5 215份山荆子叶绿体 DNA 片段的Tajima’s D值
2.2 山荆子的遗传分化
将215份山荆子按照来源省份分为7个群体(表6),对7个群体间的基因流、基因变异和分子变异进行分析。分子变异分析的结果表明遗传变异主要存在于居群内,占全部变异的93.03%,约6.97%的变异来自于居群之间,群体内和群体间可遗传变异的相关性均极显著(<0.001)(表7)。群体间3个遗传分化系数(=0.11345,=1.95;=0.06494,=3.60;=0.11353,=1.95)均不高,3个遗传分化系数体现出来的7个群体间的遗传变异分别为11.345%、6.494%和11.353%,与群体分子变异方差分析(AMOVA)中群体间方差分量的贡献率相近;而群体内的遗传变异分别为88.655%、93.506%和88.467%,都说明遗传变异主要存在于群体内部。
辽宁山荆子群体只有1份材料,因此只用剩余6个山荆子群体计算两两群体间的分化系数。6个群体两两间分化系数为-0.0247—0.216,甘肃和山西山荆子群体间遗传分化系数最低为-0.024,其次是黑龙江与吉林山荆子群体间遗传分化系数为0.002,甘肃与吉林山荆子群体间遗传分化系数最高,为0.0216。群体间遗传分化系数与其地理位置远近不完全相关(表8)。
2.3 山荆子居群间的遗传关系
计算7个山荆子群体间的遗传距离并用Neighbour-Joining(NJ)的方法构建聚类树。基于居群间遗传距离,在NJ聚类树上分成2个明显的类群。黑龙江、吉林、辽宁、内蒙古和山西的山荆子群体可归为类群Ⅰ,4个省(区)接壤连在一起,该类群分组与地理距离相关;类群Ⅱ有河北和甘肃山荆子群体,该类群分组与地理距离不相关(图2)。
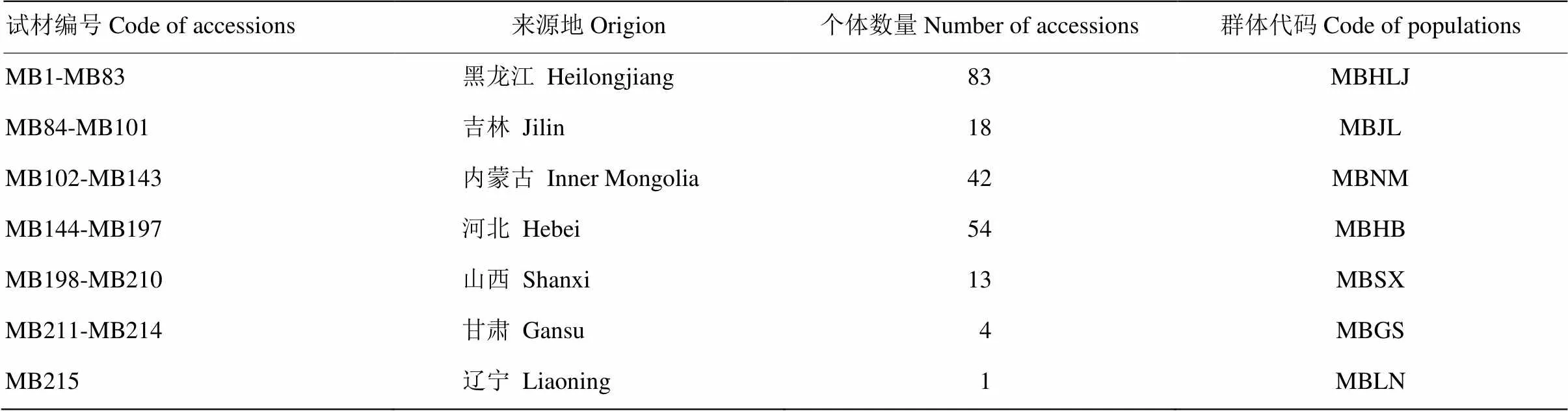
表6 215份山荆子按来源省份的7个群体
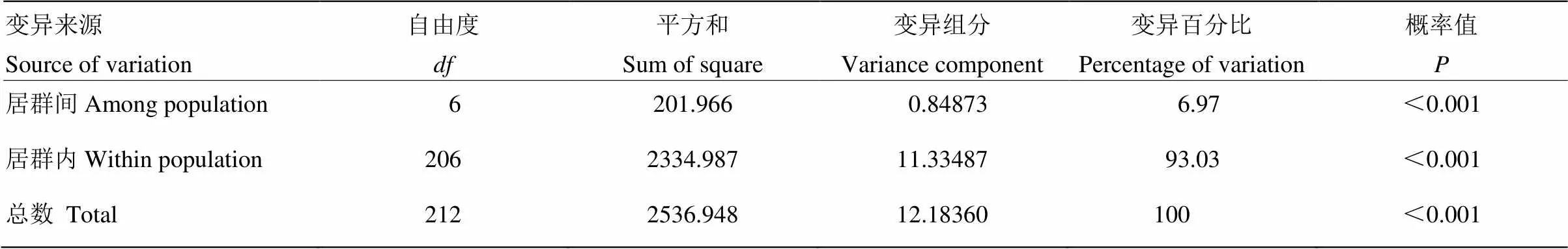
表7 山荆子居群的分子变异分析
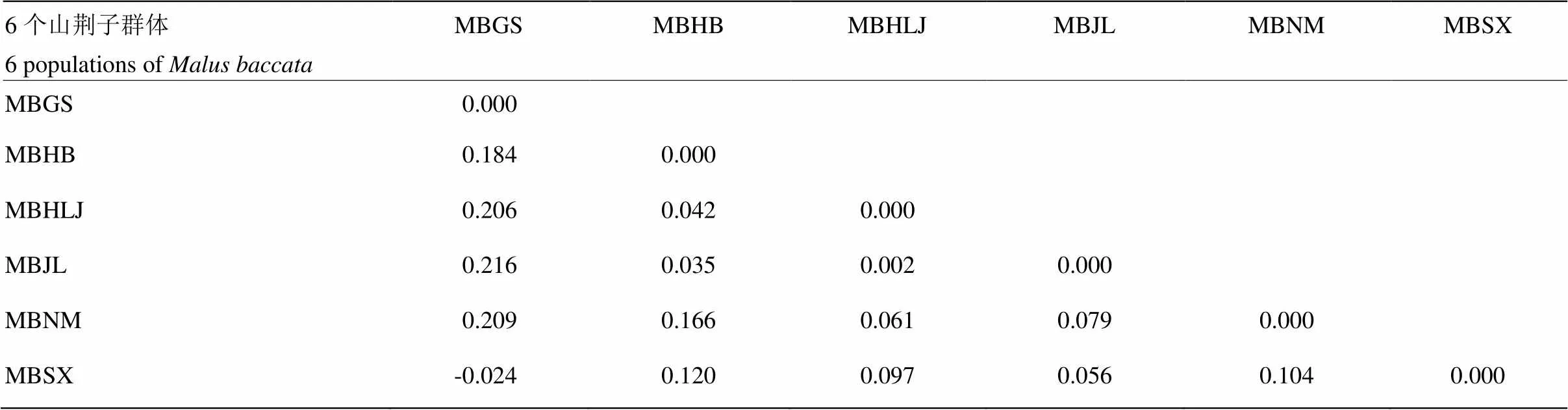
表8 6个来源地区山荆子群体间遗传分化系数
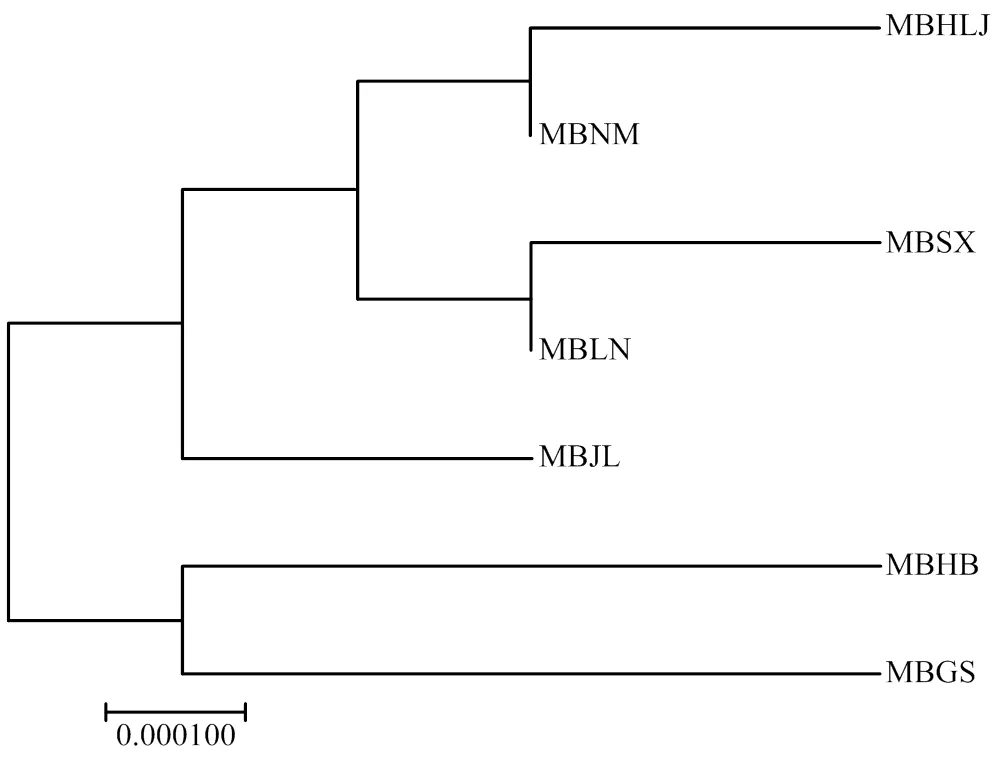
图2 基于群体间遗传距离构建的山荆子一致性NJ树
2.4 山荆子叶绿体DNA单倍型的中介网络关系
分别运用中介邻接网络算法和最大简约算法来计算和优化山荆子叶绿体DNA单倍型间的关联关系,构建苹果属山荆子种质叶绿体DNA单倍型的关联图(图3)。单倍型和中介矢量位点分别用H和mv表示。H1、H7、H21和H22位于中介网络图的躯干(Tarso)上,说明H1、H7、H21和H22分化时间要早,是较为原始的单倍型。网络图中只有一个缺失的单倍型mv1。变异位点上,H1与H7有1个位点差异,H1与H21有2个差异,H1与H22有1个差异,H7与H21有1个差异,H7与H22有2个差异,H21与H22有1个差异。网络图中H1包含H6、H13、H14、H18和H23,H16包含H17。105份山荆子种质的单倍型为H1,包含河北山荆子24份、黑龙江山荆子45份、吉林山荆子12份、辽宁山荆子1份、内蒙古山荆子14份、山西山荆子9份;3份山荆子种质的单倍型为H6,包含黑龙江山荆子2份和吉林山荆子1份;H13为1份来自吉林的山荆子,H14为1份来自内蒙古的山荆子,H18为2份来自内蒙古的山荆子,H23为来自河北的1份山荆子。H1单倍型组中包含了大部分的黑龙江、吉林、辽宁、内蒙古和山西山荆子,遗传分化较小。这进一步证实了黑龙江、吉林、辽宁、内蒙古和山西的山荆子群体遗传关系较近。
H7为2份黑龙江山荆子,H21为1份山西山荆子,H22为1份甘肃山荆子和2份河北山荆子。H3为3份黑龙江山荆子和1份内蒙古山荆子,H4为1份黑龙江山荆子和1份内蒙古山荆子,H5为8份河北山荆子和8份黑龙江山荆子,H7、H8和H9分别是2份、1份和1份黑龙江山荆子,H10包含6份河北山荆子、1份黑龙江和1份吉林山荆子,H11为1份吉林山荆子,H12为12份河北山荆子和1份吉林山荆子,H15包含的甘肃、黑龙江、内蒙古和山西山荆子分别是1份、1份、4份和3份,H16包含的甘肃、黑龙江和内蒙古山荆子分别是1份、1份和2份,H17为1份内蒙古山荆子,H19和H20分别为1份内蒙古山荆子和1份河北山荆子,H24为1份甘肃山荆子。
河北山荆子单倍型最多,有H1、H5、H10、H12、H20、H22和H23共7个单倍型;甘肃山荆子有4个单倍型,分别为H15、H16、H22和H24,H15、H16和H24均是由H22衍生的单倍型,而H22只有1份甘肃山荆子和2份河北山荆子。从叶绿体基因单倍型上,进一步证实了河北和甘肃山荆子群体的遗传关系较近。
3 讨论
山荆子叶绿体DNA多样性研究涉及到的区域有H-A、S-G spacer+intron、T-5'L和5'L-F,核苷酸多样性最高的区域为T-5'L,单倍型(基因)多样性最高的为S-G spacer+ intron,最低的为5'L-F。梨属植物叶绿体DNA间隔区D-I和L-F的多态性最高[41]。梨和苹果分别为蔷薇科的梨属和苹果属植物,其在母系遗传基因进化过程中出现了较大的差异,充分体现出非编码区适用于较低的分类阶元(如科、属)的系统研究[20]。
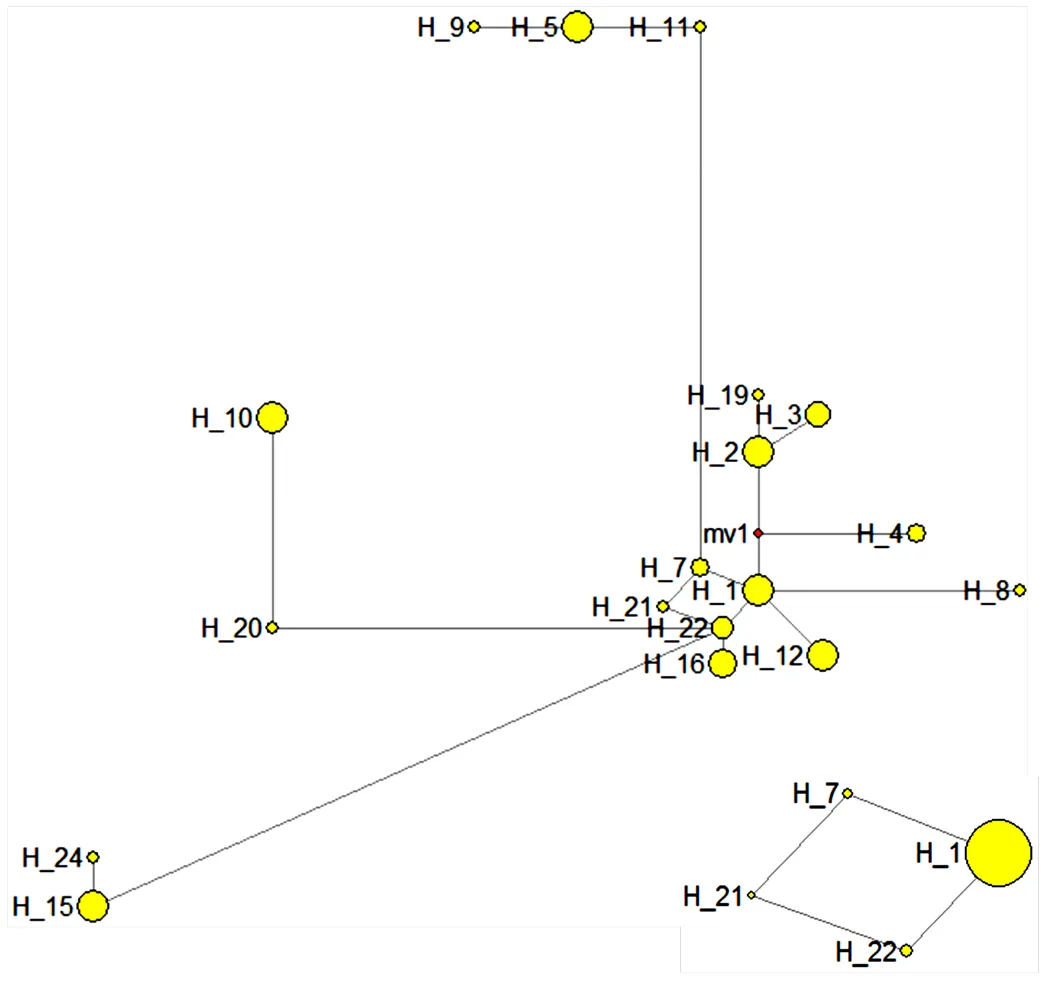
图3 基于山荆子4个叶绿体基因间区合并序列的单倍型中介邻接网络图(Median-Joining network,MJ)
叶绿体DNA的非编码区在被子植物中的遗传变异有倒位、易位、插入/缺失和核苷酸替换等[42],可利用其丰富的遗传变异有效进行亲本鉴定、遗传多样性分析以及系统发育等研究[41,43-44]。本研究4个叶绿体基因区域的突变类型主要表现为点突变、碱基插入或缺失。4区域合并后的山荆子叶绿体DNA遗传多样性(d=0.727,Pi=0.00577)高于前人在梨属豆梨[45]上鉴定的遗传多样性(d=0.719,Pi=0.00105),略低于梨属杜梨[46]的遗传多样性(d=0.807)。Tajima’s D检验中,4个cpDNA区域在各检验水平上均不显著,说明检测的4个cpDNA区域在进化上遵循中性模型,在分子水平导致进化的原因不是自然选择,而是突变压力和遗传漂变。
按照山荆子来源区域划分群体后,经过对山荆子群体的分子变异分析明确了群体内差异是遗传变异的主要来源。3个遗传分化系数中,按照NEI等[47]方法计算遗传分化系数与分子变异计算结果最接近。同时,显著大于,暗示山荆子居群中存在谱系地理结构。谱系地理探讨物种及种内居群形成当前分布格局的演化过程和历史原因,根据物种系统发育关系,结合个体地理分布,可以推测扩张、瓶颈效应、地理隔离和迁移等居群历史事件[48]。因此,对于暗示存在谱系地理结构的山荆子,基于其系统发育关系,结合其地理信息,研究其谱系地理是山荆子种质资源进一步的研究重点。
两两群体间的遗传分化系数值均小于0.05。按照WRIGHT的理论[49],值为0—0.05的群体间不存在遗传分化,值为0.05—0.15的群体间为中度遗传分化,值为0.15—0.25的群体间为高度分化。所有供试群体的值为0.11345,各群体间综合评价为中度遗传分化,表明供试山荆子的各群体曾在过去的某个时间相互发生过基因交流,但同时又抵制了基因漂变导致的群体间的遗传分化。河北和吉林山荆子群体、河北和黑龙江山荆子群体、黑龙江和吉林山荆子群体值介于0—0.05,其群体间不存在遗传分化;内蒙古和山西山荆子群体、内蒙古和吉林山荆子群体、山西和吉林山荆子群体、黑龙江和内蒙古山荆子群体、黑龙江和山西山荆子群体、河北和山西山荆子群体的值在0.15—0.25,其两两群体间为中度分化;甘肃与黑龙江山荆子群体、甘肃与吉林山荆子群体、甘肃与内蒙古山荆子群体、甘肃与河北山荆子群体、河北与内蒙古山荆子群体间的值在0.15—0.25,其群体间高度遗传分化。值越大,表明群体间遗传差异程度越大,同时群体越稳定,群体间基因交流越少;反之,值越低,表明群体间的基因交流越频繁,群体基因的稳定性就越差[50]。值通常介于0—1,而甘肃和山西山荆子群体间的遗传分化系数为负值,说明其基因交流更加频繁。除与山西山荆子群体,甘肃群体与另外4个省份的山荆子群体间均为高度的遗传分化,基因交流最少,甘肃群体最稳定。而河北与黑龙江山荆子群体间以及河北与吉林山荆子群体间的遗传分化值,仅略低于黑龙江与吉林群体间的遗传分化值。河北与黑龙江山荆子群体间以及河北与吉林山荆子群体间均无遗传分化,基因交流频繁,这与高源等[19]基于SSR对山荆子进行的群体遗传分化分析得出的结论相同。中国河北苹果栽培历史悠久,山荆子作为其中一种砧木,随苗木在各地交流频繁,供试河北山荆子主要来源于果区山地或废弃果园旁边,而黑龙江和吉林山荆子主要来源于原生境群落,推测部分河北山荆子可能主要来源于黑龙江和吉林地区。群体间基于遗传距离的聚类分析表明,黑龙江、吉林、辽宁、内蒙古和山西山荆子群体间的遗传距离相对较近,而甘肃与河北山荆子群体间遗传距离较近。
所有山荆子种质共有24个单倍型,有4个单倍型位于中介邻接网络的躯干上,其中单倍型H1包含了6个来源地区的105份山荆子种质,是所有单倍型中种质数量最多、来源最广泛的。若单倍型在种群或居群中覆盖范围最广、出现频率最高,并处于单倍型网络的中心位置,那么这种单倍型很可能是网络图的所有单倍型中最为古老的类型[51]。因此,H1单倍型可以被认为是最古老的单倍型。以H1为中心呈星状辐射,说明其发生种群扩张。位于躯干上的闭合环所包含的4个单倍型涵盖了除甘肃外的所有来源地区,而处于邻接网络外围的单倍型也涵盖了所有来源地区。
不同群体种质数量的差异可能会影响群体内的遗传多样性水平和群体间的遗传分化系数,虽然在遗传分化的分析过程中去除了仅有1份种质的辽宁山荆子群体,但不同来源的山荆子群体之间的遗传关系和遗传分化不完全与地理距离相关的情况,有可能是不同来源的群体数量差异造成的。因此,还要继续加大种质资源收集的力度,对于群体数量比较少的区域,进一步细化收集区域,目标是即使以少量的种质也能代表该区域的遗传变异;对于群体数量比较多的区域,扩大考察范围,可能会有新的叶绿体基因单倍型出现,为苹果属不同种以及种下类型的起源和演化提供新的证据。
4 结论
山荆子可能为多点起源。结合山荆子遗传分化和遗传关系的分析,推测黑龙江和吉林为一个起源地,地域跨度大的内蒙古为一个起源地,甘肃和山西为一个起源地,3个区域分别由大兴安岭和阴山山脉分隔;河北地区山荆子含有最古老的单倍型,但受人为散播的影响,其为初生分布中心还是次生分布中心有待进一步研究。各个起源地区的山荆子经历着种群扩张的进化过程,同时作为砧木在各地区间交流频繁程度的不同造成各地区间山荆子遗传分化的差异。
[1] 中国植物志编辑委员会. 中国植物志[DB/OL]. [2019-05-17]. http://frps.iplant.cn/.
Editorial Committee of Flora Reipublicae Sinicae.[DB/OL]. [ 2019-05-17]. http://frps.iplant.cn/. (in Chinese)
[2] 俞德俊. 中国果树分类学. 北京: 中国农业出版社, 1979: 91.
YU D J.. Beijing: China Agriculture Press, 1979: 91. (in Chinese)
[3] 钱关泽. 苹果属 (Mill.) 分类学研究[D]. 南京: 南京林业大学, 2005.
QIAN G Z. The taxonomic study of the genusMill. [D]. Nanjing: Nanjing Forestry University, 2005. (in Chinese)
[4] ELITH J, GRAHAM C H, ANDERSON R P, DUDíK M, FERRIER S, GUISAN A, HIJMANS R J, HUETTMANN F, LEATHWICK J R, LEHMANN A, LI J, Lohmann L G, LOISELLE B A, MANION G, MORITZ C, NAKAMURA M, NAKAZAWA Y, OVERTON J M, PETERSON A T, PHILLIPS S J, RICHARDSON K, SCACCHETTI- PEREIRA R, SCHAPIRE R E, SOBERON J, WILLIAMS S, WISZ M S, ZIMMERMANN N E. Novel methods improve prediction of species’ distributions from occurrence data., 2006, 29(2): 129-151.
[5] 王大江, 王昆, 高源, 赵继荣, 刘立军, 龚欣, 李连文. 我国苹果属资源现代分布调查初报. 植物遗传资源学报, 2017, 18(6): 1116-1124.
WANG D J, WANG K, GAO Y, ZHAO J R, LIU L J, GONG X, LI L W. Preliminary investigation of modern distribution ofresources in China., 2017, 18(6): 1116-1124. (in Chinese)
[6] 杜学梅, 杨廷桢, 高敬东, 王骞, 蔡华成, 李春燕, 弓桂花. 中国野生山定子的自然分布及利用研究现状. 中国农学通报, 2017, 33(5): 24-28.
DU X M, YANG T Z, GAO J D, WANG Q, CAI H C, LI C Y, GONG G H. Wild(L.) Borkh. in China: Natural distribution, utilization and study status., 2017, 33(5): 24-28. (in Chinese)
[7] 王雷宏. 山荆子((L.) Borkh.)变异式样研究[D]. 南京: 南京林业大学, 2008.
WANG L H. A study on variation patterns in(L.) Borkh. [D]. Nanjing: Nanjing Forestry University, 2008. (in Chinese)
[8] 陆秋农, 贾定贤. 中国果树志•苹果卷. 北京: 中国林业出版社, 1999: 121.
LU Q N, JIA D X.•. Beijing: China Forestry Press, 1999: 121. (in Chinese)
[9] 李育农. 苹果属植物种质资源研究. 北京: 中国农业出版社, 2001: 23-25.
LI Y N.ofMill.. Beijing: China Agriculture Press, 2001: 23-25. (in Chinese)
[10] 邹旭, 彭冶, 王璐, 李垚, 张往祥, 刘雪. 末次盛冰期以来气候变化对中国山荆子分布格局的影响. 植物科学学报, 2018, 36(5): 676-686.
ZOU X, PENG Y, WANG L, LI Y, ZHANG W X, LIU X. Impact of climate change on the distribution pattern of(L.) Borkh. in China since the Last Glacial Maximum., 2018, 36(5): 676-686. (in Chinese)
[11] 王雷宏, 汤庚国. 山荆子腊叶标本表型性状变异分析. 西北植物学报, 2007, 27(8): 1690-1694.
WANG L H, TANG G G. Phenotypic variations of exsiccate- specimen of(L.) Borkh., 2007, 27(8): 1690-1694. (in Chinese)
[12] 王雷宏, 汤庚国, 夏海武, 蔡华. 山荆子叶脉序的研究. 南京林业大学学报(自然科学版), 2008, 32(2): 39-42.
WANG L H, TANG G G, XIA H W, CAI H. Study on leaf venation of(L.) Borkh.., 2008, 32(2): 39-42. (in Chinese)
[13] 陈曦, 汤庚国, 郑玉红, 王雷宏. 苹果属山荆子遗传多样性的RAPD 分析. 西北植物学报, 2008, 28(10): 1954-1959.
CHEN X, TANG G G, ZHENG Y H, WANG L H. RAPD analysis of genetic diversity of(L.) Borkh., 2008, 28(10): 1954-1959. (in Chinese)
[14] ROBINSON J P, HARRIS S A, JUNIPER B E. Taxonomy of the genusMill. ( Rosaceae ) with emphasis on the cultivated apple,Borkh., 2001, 226(1/2): 35-58.
[15] 王雷宏, 杨俊仙, 郑玉红, 汤庚国. 苹果属山荆子地理分布模拟. 北京林业大学学报, 2011, 33(3): 70-73.
WANG L H, YANG J X, ZHENG Y H, TANG G G. Modelling the geographical distribution of., 2011, 33(3): 70-73. (in Chinese)
[16] 杨锋, 刘志, 伊凯, 刘延杰, 王强, 孙建设. 东北山定子((L.) Borkh.)野生居群表型遗传多样性分析及生态地理分布研究. 植物遗传资源学报, 2015, 16(3): 490-496.
YANG F, LIU Z, YI K, LIU Y J, WANG Q, SUN J S. Studies on geographical regions and analysis on genetic diversity of phenotypic of natural population of ‘( L.) Borkh’ in Northeast of China., 2015, 16(3): 490-496. (in Chinese)
[17] 王雷宏, 郑玉红, 汤庚国. 8个山荆子居群遗传多样性的ISSR分析. 西北植物学报, 2010, 30(7): 1337-1343.
WANG L H, ZHENG Y H, TANG G G. ISSR analysis of genetic diversity of eight populations in., 2010, 30(7): 1337-1343. (in Chinese)
[18] 王雷宏, 郑玉红, 汤庚国. 基于SSR标记的8个山荆子居群遗传多样性和遗传关系分析. 植物资源与环境学报, 2012, 21(1): 42-46.
WANG L H, ZHENG Y H, TANG G G. Analyses of genetic diversity and genetic relationship of eight populations ofbased on SSR marker., 2012, 21(1): 42-46. (in Chinese)
[19] 高源, 王昆, 王大江, 赵继荣, 张彩霞, 丛佩华, 刘立军, 李连文, 朴继成. 7 个来源地区山荆子的遗传多样性与群体结构分析. 中国农业科学, 2018, 51(19): 3766-3777.
GAO Y, WANG K, WANG D J, ZHAO J R, ZHANG C X, CONG P H, LIU L J, LI L W, PIAO J C. The genetic diversity and population structure analysis on(L.) Borkh. from 7 sources., 2018, 51(19): 3766-3777. (in Chinese)
[20] 倪梁红, 赵志礼, 米玛. 药用植物叶绿体基因组研究进展. 中药材, 2015, 38(9): 1990-1994.
NI L H, ZHAO Z L, MI M. Advances in research on chloroplast genome of medicinal plants., 2015, 38(9): 1990-1994. (in Chinese)
[21] 李宏韬, 赵淑青, 赵彦修, 张慧. 叶绿体基因工程简介. 遗传, 2003, 25(4): 495-498.
LI H T, ZHAO S Q, ZHAO X X, ZHANG H. The introduction of chloroplast gene engineering., 2003, 25(4): 495-498. (in Chinese)
[22] 张韵洁, 李德铢. 叶绿体系统发育基因组学的研究进展. 植物分类与资源学报, 2011, 33(4): 365-375.
ZHANG Y J, ZHANG D Z. Advances in phylogenomics based on complete chloroplast genomes., 2011, 33(4): 365-375. (in Chinese)
[23] 付涛, 王志龙, 钱萍仙, 李文, 袁冬明, 严春风. 高等植物DNA条形码最新研究进展及其应用. 核农学报, 2016, 30(5): 887-896.
FU T, WANG Z L, QIAN P X, LI W, YUAN D M, YAN C F. The latest research progress and application of the DNA barcode in higher plants., 2016, 30(5): 887-896. (in Chinese)
[24] SAVOLAINEN V, CORBAZ R, MONCOUSIN C, SPICHIPER R, MANEN J F. Chloroplast DNA variation and parentage analysis in 55 apples., 1995, 90: 1138-1141.
[25] ROBINSON J P, HARRIS S A, JUNIPER B E. Taxonomy of the genusMill. () with emphasis on the cultivated apple,Borkh., 2001, 226: 35-58.
[26] COART E, VAN GLABEKE S, DE LOOSE M, LARSEN A S, ROLDAN-RUIZ I. Chloroplast diversity in the genus: new insights into the relationship between the European wild apple ((L) Mill.) and the domesticated apple (Borkh.)., 2006, 15: 2171-2182.
[27] NIKIFOROVA S V, CAVALIERI D, VELASCO R, GOREMYKIN V. Phylogenetic analysis of 47 chloroplast genomes clarifies the contribution of wild species to the domesticated apple maternal line., 2013, 30(8): 1751-1760.
[28] 丁芳兵. 湖北海棠()及近缘种的K和ITS序列分析[D]. 南京: 南京林业大学, 2012.
DING F B. Sequence analysis ofK and ITS betweenand its relative species [D]. Nanjing: Nanjing Forestry University, 2012. (in Chinese)
[29] 朱元娣, 曹敏格, 许正, 王昆, 张文. 基于ITS和K序列探讨新疆野苹果与中国苹果的系统演化关系. 园艺学报, 2014, 41(2): 227-239.
ZHU Y D, CAO M G, XU Z, WANG K, ZHANG W. Phylogenetic relationship between Xinjiang wild apple(Roem.)and Chinese apple(×subsp.)based on ITS andK sequences., 2014, 41(2): 227-239. (in Chinese)
[30] 徐榕雪. 基于分子标记技术的部分苹果属植物分类地位及亲缘关系研究[D].南京: 南京林业大学, 2018.
XU R X. Taxonomic status and phylogenetic relationships of someplants based on molecular marker technology [D]. Nanjing: Nanjing Forestry University, 2018. (in Chinese)
[31] 李慧峰. 泰沂山区苹果属植物系统学研究[D]. 沈阳: 沈阳农业大学, 2012.
LI H F. Studies on the taxonomy of the genusMill. (Rosaceae) of Taiyi-Mountains [D]. Shenyang: Shenyang Agricultural University, 2012. (in Chinese)
[32] BARAKET G, OLFA S, KHALED C, MESSAOUD M, MOHAMED M, MOKHTAR T, AMEL S H. Chloroplast DNA analysis in Tunisian fig cultivars (L.): Sequence variations of theintergenic spacer., 2009, 36(11): 828-835.
[33] POTTER D, LUBY J J, HARRISON R E. Phylogenetic relationships among species of(Rosaceae) inferred from non-coding nuclear and chloroplast DNA sequences., 2009, 25(2): 337-348.
[34] GHADA B, AHMED B A, KHALED C, OLFA S, MESSAOUD M, MOKHTAR T, AMEL S H. Molecular evolution of chloroplast DNA in fig (L.): Footprints of sweep selection and recent expansion., 2010, 38(4): 563-575.
[35] SHAW J, LICKEY E B, BECK J T, FARMER S B, LIU W S, MILLER J, SIRIPUN K C, WINDER C T, SCHILLING E E, SMALL R L. The tortoise and the hare Ⅱ: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis., 2005, 921(1): 142-166.
[36] VOLK G M, HENK A D, BALDO A, FAZIO G, CHAO C T, RICHARDS C M. Chloroplast heterogeneity and historical admixture within the genus., 2015, 102(7): 1198-1208.
[37] KUMAR S, STECHER G, TAMURA K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets., 2016, 33(7): 1870-1874.
[38] LIBRADO P, ROZAS J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data., 2009, 25: 1451-1452.
[39] SAITO N, NEI M. The neighbor-joining method: A new method for reconstructing phylogenetic trees., 1987, 4(4): 406-425.
[40] EXCOFFIER L, LISCHER H E L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows., 2010, 10: 564-567.
[41] KIMURA T, IKETANI H, KOTOBUKI K, MATSUTA N, BAN Y, HAYASHI T, YAMAMOTO T. Genetic characterization of pear varieties revealed by chloroplast DNA sequences., 2003, 7: 241-247.
[42] PETIT R,Aguinagalde I, de Beaulieu J, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Müller-Starck G, Demesure- Musch B, Palmé A, Martín J P, Rendell S, Vendramin G G. Glacial refugia: Hotspots but not melting pots of genetic diversity., 2003, 300: 1563-1565.
[43] KATAYAMA H, OGIHARA Y. Phylogenetic affinities of the grasses to other monocots as revealed by molecular analysis of chloroplast DNA., 1996, 20: 527-581.
[44] EIADTHONG W, YONEMORI K, SUGIURA A, UTSUNOMIYA N, SUBHADRABANDHU S. Analysis of phylogenetic relationships in Mangifera by restriction site analysis of an amplified region of cpDNA., 1999, 80: 145-150.
[45] LIU J, ZHENG X Y, DANEIL P, Hu C Y, TENG Y W. Genetic diversity and population structure of(Rosaceae) in Zhejiang province, China., 2012, 45: 69-78.
[46] 宗宇. 中国北方野生杜梨的遗传多样性和谱系地理研究[D]. 杭州: 浙江大学, 2014.
ZONG Y. Studies on genetic diversity and phylogeography of wildin Northern China[D]. Hangzhou: Zhejiang University, 2014. (in Chinese)
[47] NEI M. Estimation of average heterozygosity and genetic distance from a small number of individuals., 1978, 89(3): 583-590.
[48] AVISE J C, ARNOLD J, BALL R M, BERMINGHAM E, LAMB T, NEIGEL J E, REEB C A, SAUNDER N C. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics., 1987, 18: 489-522.
[49] WRIGHT S.. Chicago: The University of Chicago Press, 1978: 4.
[50] ZHANG C Y, CHEN X S, HE T M, LIU X L, FENG T, YUAN Z H. Genetic structure ofpopulation from Xinjiang, China, revealed by SSR markers., 2007, 34(10): 947-955.
[51] POSADA D, CRANDALL K A. Intraspecific gene genealogies: Trees grafting into networks., 2001, 16: 37-45.
Genetic Diversity and Phylogenetics of(L.) Borkh Revealed by Chloroplast DNA Variation
GAO Yuan, WANG DaJiang, WANG Kun, CONG PeiHua, ZHANG CaiXia, LI LianWen, PIAO JiCheng
(Research Institute of Pomology, Chinese Academy of Agricultural Sciences/Key Laboratory of Horticultural Crops Germplasm Resources Utilization, Ministry of Agriculture, Xingcheng 125100, Liaoning)
【Objective】(L.) Borkh is the most widely distributed native species in China. The non-coding region of chloroplast genome by maternal inheritance is suitable for the systematic study of lower taxonomic levels (such as families and genera). The non-coding regions of cpDNA of 215 germplasms were sequenced, and their genetic variation was analyzed. In this study, the genetic diversity ofand the phylogenetic relationship among different populations were explored from the perspective of maternal inheritance, which provided a theoretical basis for origin and genetic evolution, collection and protection ofgermplasm resources in China.【Method】Four non-coding regions (H-A,S-G spacer + intron,T-5'L and 5'L-F) of 215 germplasms were amplified by four primers. After manually proofreading sequences obtained through forward and backward sequencing, MEGA 7.0 was used for sequence splicing and alignment, and based on genetic distance, the Neighbour-Joining phylogenetic tree was constructed among different populations of. DnaSP ver5.10.01 was used to calculate the genetic diversity parameters of chloroplast DNA, gene flow and gene differentiation among different populations. Arlequin v3.5 was used to analyze standard molecular variation (AMOVA), and NetWork 4.6.1.2 was used to construct Median-Joining network for cpDNA haplotypes among intraspecific populations of.【Result】The length of four non-coding regions of chloroplast DNA was 3 777 bp after sequencing, splicing, alignment and merging, and 171 variable sites were detected, which included 1 singleton variable sites, 20 parsimony informative sites and 150 insertion-deletion gaps. Among 215 accessions of, the number of variable sites of regionH-A,S-G spacer + intron,T-5'L and 5'L-F were 26, 32, 103 and 10, respectively. The number of haplotypes for four regions were 8, 8, 6 and 4, respectively, and after four regions merged, the haplotypes of chloroplast DNA fragments were 24. The region with highest nucleotide diversity wasT-5'L (Pi=0.01174), and the region with highest haplotype diversity wasS-G spacer + intron (d=0.599), and the haplotype diversity of 5'L-F was the lowest (d=0.228). The cpDNA diversity ofwas high (d=0.727, Pi=0.00577).Tajima’s test showed all Tajima’s D values were not statistical at different levels, which indicated that variation of those chloroplast regions followed natural theory of molecular evolution. AMOVA showed that genetic variation mainly existed within populations.【Conclusion】The four non-coding regions of chloroplast DNA were suitable for the analysis of genetic diversity and phylogenetics of. At the cpDNA level, it was not natural selection, but mutation pressure and genetic drift that led to population evolution of. The genetic differentiation among populations was not completely correlated with their geographical distance.might originate from several sites, and the three possible origins, including Heilongjiang and Jilin, Inner Mongolia, Gansu and Shanxi, were inferred.
; chloroplast DNA; non-coding region; genetic diversity; phylogenetics
2019-07-21;
2019-11-01
国家公益性行业(农业)科研专项(201303093)、中国农业科学院创新工程项目(CAAS-ASTIP-2018-RIP-02)、农作物种质资源保护(NB2015-2130135-39)
高源,E-mail:gaoyuan02@caas.cn。通信000作者王昆,E-mail:wangkun5488@163.com。通信作者丛佩华,E-mail:congph@163.com
(责任编辑 赵伶俐)
猜你喜欢
今日农业(2022年14期)2022-09-15
今日农业(2022年13期)2022-09-15
清华金融评论(2022年4期)2022-04-13
中国种业(2022年1期)2022-01-27
国际放射医学核医学杂志(2021年10期)2021-02-28
房地产导刊(2020年7期)2020-08-24
四川蚕业(2020年4期)2020-02-10
中国外汇(2019年14期)2019-10-14
课程教育研究·学法教法研究(2019年18期)2019-10-08
少儿科技(2019年9期)2019-09-10
