
二倍体和四倍体西瓜的转录组初步分析
2020-02-22 07:42许哲张晨光张思远王先裕高建昌
江苏农业科学 2020年24期
许哲 张晨光 张思远 王先裕 高建昌
摘要:植物染色体加倍后的表型变化与基因的表达密切相关。本研究对1份二倍体和3份四倍体西瓜材料进行转录组测序并分析差异表达基因。结果表明,二倍体和四倍体西瓜的差异表达基因显著富集的GO term為细胞氮化合物生物合成过程、光系统、半胱氨酸型内肽酶抑制剂活性、氧化还原酶活性以及类囊体等,KEGG富集分析中发现差异表达基因在光合作用、光合作用天线蛋白、叶绿素和卟啉代谢、乙醛酸和二羧酸代谢等通路较为活跃。二倍体和四倍体西瓜中高水平差异基因大都是上调表达,氧化应激蛋白、脱落酸受体、乙烯响应转录因子等差异表达基因可能是西瓜多倍体形成的关键因子,对相关基因的深入研究有助于解析植物倍性变化的分子机制。
关键词:多倍体;西瓜;转录组;差异表达基因;GO富集分析;KEGG富集分析
中图分类号: S651.01 文献标志码: A 文章编号:1002-1302(2020)24-0053-07
西瓜(Citrullus lanatus)原产于非洲,属于葫芦科西瓜属,为一年生蔓生藤本植物,是世界上重要的园艺作物。我国是世界上西瓜生产与消费大国,2016年西瓜播种面积为189.08万hm2,总产量为7 940万t[1]。
四倍体西瓜是普通二倍体西瓜染色体加倍的结果,在田间其最明显的特征是巨大性,叶片大且肥厚,茎粗且节间短,叶色浓绿,花和果实也较为巨大,综合抗病能力较强。染色体加倍获得四倍体是选育三倍体西瓜(无籽西瓜)的基础。我国从20世纪50年代开始对多倍体西瓜诱导方法的研究,如今已建立秋水仙素诱导、体细胞杂交、胚乳培养等多种四倍体诱导方法;而且建立了染色体计数、叶肉细胞分析、流式细胞仪计数等染色体倍性检测方法。利用上述方法选育出很多无籽西瓜品种,为我国西瓜产业的发展做出了巨大贡献。但是,对四倍体西瓜表型变化与染色体倍性的关系还缺乏分子层面的认知。
转录组是研究细胞表型和功能的一个重要手段,是指某一特定的生理条件下,细胞、组织或生物体内所有转录产物的集合,即转录后所有mRNA的总称[2]。对二倍体和四倍体西瓜进行转录组水平的分析,为解析四倍体表型的成因提供了新的手段。自2012年西瓜全基因组测序完成后,西瓜转录组分析亦成为研究热点。Wechter等利用832个表达序列标签研究了果实发育过程中的基因表达情况[3];Guo等在西瓜果实发育和成熟过程中鉴定出3 023个差异表达基因[4];Zhu等通过比较2个红色和黄色果肉的栽培品种,确定了797个新基因[5];龙娅丽等通过研究二倍体西瓜及其同源四倍体叶片sRNA表达谱,提供了多倍体抗逆性强的理论依据[6]。
多倍体化是植物进化中的一种普遍现象,也是新物种形成的重要途径[7]。目前研究者多集中在对二倍体西瓜的研究,关于二倍体西瓜与四倍体西瓜转录组差异的研究相对较少。本研究利用Illumina二代高通量测序平台对二倍体西瓜以及3份四倍体西瓜叶片进行转录组测序分析,比较二倍体西瓜和四倍体西瓜中基因表达的差异,以期通过转录组测序(RNA-Seq)技术获得与倍性相关的基因序列,为揭示西瓜的倍性机制提供参考。
1 材料与方法
1.1 试验材料
试验材料为1份二倍体,记为WMA,3份四倍体,记为WMB、WMC和WMD。WMB为WMA染色体加倍获得,为了增加样品的遗传多样性,加入另外2份不同来源的四倍体材料WMC、WMD,材料均来自中国农业科学院蔬菜花卉研究所西瓜育种课题组。本试验未设生物学重复。试验地点为中国农业科学院蔬菜花卉研究所日光温室,2019年6月1日将西瓜种子播种于穴盘中,20 d后长出真叶时取2 cm2大小新鲜叶片,立即放入液氮中保存备用。
1.2 总RNA的提取及检测
使用天根生化科技(北京)有限公司RNA试剂盒对试验材料进行总RNA提取,后通过Nanodrop检测RNA纯度(D260 nm/280 nm、D260 nm/230 nm),通过Agilent 2100对RNA片段长度进行检测。
1.3 cDNA文库构建和测序
样品检测合格后,使用带有Oligo(dT)的磁珠富集mRNA,之后加入破碎缓冲液将mRNA打断成短片段。再用六碱基随机引物(random hexamers)以mRNA为模板进行反转录合成一链cDNA,加入缓冲液、dNTPs和DNA聚合酶Ⅰ合成二链cDNA。接着,利用AMPure XP beads纯化双链cDNA。对纯化后的双链cDNA进行末端修复、加A(特异碱基)、加接头。通过AMPure XP beads核酸纯化试剂盒对双链cDNA进行片段大小选择,最后进行PCR扩增以构建cDNA文库。
文库构建完成后用Agilent 2100对文库的插入片段大小进行检测,当片段大小符合预期时,再使用定量PCR(Q-PCR)方法对文库的有效浓度进行精确定量(文库有效浓度>4 nmol/L),以达到高质量文库标准,最后通过Illumina二代高通量测序平台,采用PE150双末端测序策略,完成RNA-Seq。
1.4 数据质控与基因功能注释
下机数据(raw data)通常会含有少量的接头污染及低质量的Reads,如果不对其进行过滤处理会对后续分析造成影响,为此我们过滤掉了带有测序接头adapter的Reads、N(不确定碱基)含量比例大于10%的Reads以及低质量碱基(Q≤20)含量大于50%的Reads。使用TopHat2软件将数据过滤后的Clean Reads与西瓜参考基因组[8]进行序列比对,之后用DEGseq[9]进行差异表达分析,再分别与基因本体(GO)、京都基因与基因组百科全书(KEGG)数据库比对,获得相关注释信息。
2 结果与分析
2.1 转录组测序数据质量评估
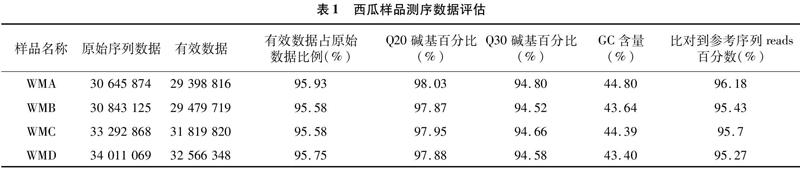
将二倍体西瓜与四倍体西瓜叶片RNA样本进行测序,结果见表1。二倍体WMA和四倍体WMB、WMC以及WMD有效数据均占原始序列数据的9558%以上,Q20(碱基识别错误率为1%)碱基百分比均大于97.87%,Q30(碱基识别错误率为01%)碱基百分比均大于94.52%,GC含量均大于43.40%,能比对到参考序列的reads百分数均大于95.27%,说明测序结果有较高的准确率。
2.2 RNA-Seq相关性检查
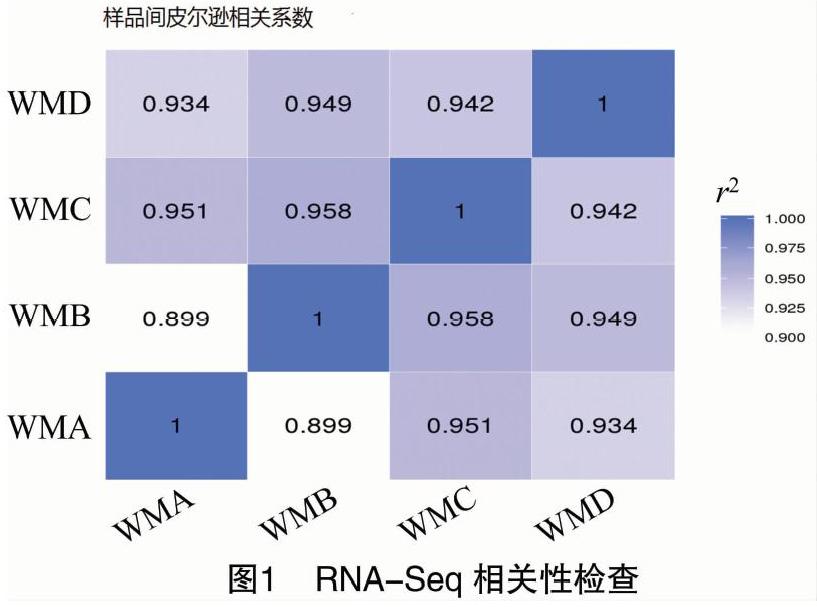
为了检测转录组测序的相关性,对每个样品进行相关性分析。相关性分析是基于样品间基因整体的表达水平做的皮尔逊相关系数分析,相关系数越接近1,表明样品之间表达模式的相似度越高。Encode计划建议皮尔逊相关系数的平方(r2)大于0.92(理想的取样和试验条件下)。而实际项目操作中,要求r2至少要大于0.8,否则须要对样品做出合理的解释,或重新进行试验。本次试验中WMA、WMB、WMC和WMD 4个样品之间相关系数r2均大于0.899,最高达到了0.958(图1),表明样品间相关性较好。
2.3 不同倍性西瓜差异表达基因分析
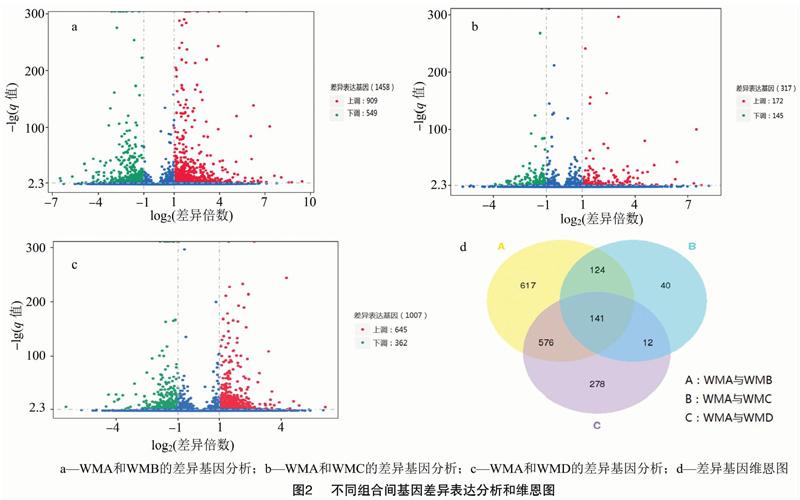
2.3.1 二倍体WMA与四倍体WMB、WMC和WMD差异表达基因筛选 对于无生物学重复的试验,为避免引入试验误差,应该对结果进行严格控制,对差异基因进行筛选的阈值一般为:|log2(差异倍数)|>1 且q值<0.005[10]。结果(图2)表明,在WMA和WMB中共检测到1 458条差异表达基因,其中表达上调的基因有909条,表达下调的基因有549条;在WMA和WMC中共检测到317条差异表达基因,其中表达上调的基因有172条,表达下调的基因有145条;在WMA和WMD中共检测到 1 007 条差异表达基因,其中表达上调的基因有645条,表达下调的基因有362条。不同组间差异基因维恩图显示,有141个不同组间的共同差异表达基因。
2.3.2 差异基因的聚类分析 从图3可以看出,WMB和WMD高表达量和低表达量基因模式大体相似。WMA、WMB、WMC和WMD的差异表达聚合为5类。A组中,WMA和WMD基因表达量以上调为主,WMB和WMC基因表达量以下调为主;B组中,WMA基因表达量呈下调趋势,WMB、WMC和WMD基因表达量以下调为主;C组中WMA和WMC基因表达量以上调为主,WMB和WMD基因表达量以下调为主;D组中,WMA和WMC基因表达量以下调为主,WMB和WMD基因表达量以上调为主;E组中,WMA、WMB、WMC和WMD基因表达量各不一致。二倍體WMA和四倍体WMB、WMD之间的差异表达基因主要分布在C和D组中,这部分可能调控西瓜倍性的差异基因。
2.3.3 差异基因GO富集分析 GO是一个用于描述生物体中基因和蛋白质的功能分类体系。GO分为分子功能(molecular function)、生物过程(biological process)、和细胞组成(cellular component)等3个方面。GO的基本单位为term,每个term对应一个功能或属性。在分析中采用的软件为GOseq[11],前30个富集最显著的GO term见图4,如果不足30个,则全部展示。结果表明,WMA和WMB之间富集的差异表达基因与生物过程、细胞组成和分子功能相关的分别有21、3、6个GO term。说明大部分差异基因与生物过程相关,其中与生物过程相关富集最显著、基因数量最多的term为小分子代谢过程(small molecule metabolic process)和细胞氮化合物生物合成过程(cellular nitrogen compound biosynthetic process);与细胞组成相关,富集最显著的term为光系统Ⅰ(photosystem Ⅰ),富集基因数量最多的类别为光系统(photosystem);与分子功能相关富集最显著的term为半胱氨酸型内肽酶抑制剂活性(cysteine-type endopeptidase inhibitor activity),富集基因数量最多的term为氧化还原酶活性(oxidoreductase activity)。
在WMA和WMC之间,富集的差异表达基因与生物过程、细胞组成和分子功能相关的分别有15、0、15个GO term。其中与生物过程相关富集最显著的term为发病机制(pathogenesis),富集基因数量最多的term为胺生物合成过程(amine biosynthetic process);与分子功能相关富集最显著、基因数量最多的term为内肽酶抑制剂活性(endopeptidase inhibitor activity)、内肽酶调节剂活性(endopeptidase regulator activity)、肽酶抑制剂活性(peptidase inhibitor activity)、肽酶调节剂活性和酶抑制剂活性(enzyme inhibitor activity)。
在WMA和WMD之间,富集的差异表达基因与生物过程、细胞组成和分子功能相关的分别有19、5、6个GO term。其中与生物过程相关富集最显著、基因数量最多的term为电子转运(electron transport),其次为细胞氮化合物生物合成过程(cellular nitrogen compound biosynthetic process);与细胞组成相关,富集最显著、基因数量最多的term为类囊体(thylakoid)和类囊体组分(thylakoid part);与分子功能相关富集最显著、基因数量最多的term为氧化还原酶活性(oxidoreductase activity)。
2.3.4 差异基因的KEGG注释 KEGG是一个整合了基因组、化学和系统功能信息的数据库,是系统分析基因产物在细胞中的代谢途径以及基因产物功能的数据库[12]。本研究以KEGG代谢途径数据库为依据[13],差异表达基因显著富集的前20条通路见图5。在WMA和WMB之间,差异表达基因富集最显著的途径有光合作用、光合作用天线蛋白、乙醛酸和二羧酸代谢、卟啉和叶绿素代谢等;在WMA和WMC之间,差异表达基因富集最显著的途径有异喹啉生物碱生物合成、牛磺酸和亚牛磺酸代谢、托烷和哌啶、吡啶生物碱的生物合成等;在WMA和WMD之间,差异表达基因富集最显著的途径有光合作用天线蛋白、光合作用、卟啉和叶绿素代谢、乙醛酸和二羧酸代谢等。
3 结论与讨论
多倍体化后,随着染色体的加倍,染色体上每个位点等位基因的数量也会发生增倍,并可能由此导致基因表达质和量的变化[7]。本试验中,通过对二倍体西瓜和四倍体西瓜的差异表达基因进行GO功能注释,主要GO term为细胞氮化合物生物合成过程、光系统、半胱氨酸型内肽酶抑制剂活性、氧化还原酶活性以及类囊体。Compton等认为叶长与叶宽是西瓜倍性水平很好的标志,四倍体子房直径是二倍体子房直径的1.4倍[14]。蔡力研究发现,同一发育时期四倍体紫锥菊叶绿素a、叶绿素b和总叶绿素含量均高于二倍体紫锥菊[15]。本次试验发现的差异表达基因大都与植株的光合作用有关,一定程度上验证、解释了前人的研究,说明四倍体植株形成需要调控光合作用的相关基因有较高的表达量。
通过KEGG显著性富集分析发现差异表达基因主要在光合作用、光合作用天线蛋白、叶绿素和卟啉代谢、乙醛酸和二羧酸代谢等通路中较为活跃,说明它们在四倍体植株形成和生长过程中发挥重要作用。
多倍体化不仅导致植物基因组大小以及结构发生改变,还影响了基因的表达[16]。为了挖掘在二倍体和四倍体西瓜中与倍性相关的基因,笔者查找了几个显著富集的代谢通路下的差异表达基因,结合基因定量FPKM,发现一些在四倍体中表达量明显高于二倍体的基因:Cla97C10G205730、Cla97C10G187010、Cla97C01G019450、Cla97C02G026280、Cla97C01G004920和Cla97C07G140200,并对它们进行了GO功能注释。
Cla97C10G187010、Cla97C02G026280、Cla97C01G004920、Cla97C07G140200是调控细胞核成分的基因,控制多倍体西瓜细胞遗传与代谢。研究发现西瓜四倍体叶片比二倍体的叶片大而且厚,颜色也较深[17],Cla97C10G205730和Cla97C01G019450是调控西瓜细胞壁、液泡膜、内质网、高尔基体等膜结构的基因,它们在四倍体西瓜中的表达量上调较为明显,促进了细胞中膜结构的发育,可能是多倍体西瓜叶片大而厚的主要原因。
另外,对3组对比中差异倍数达到2倍以上的差异表达基因进行了注释分析。结果表明二倍体和四倍体西瓜中高水平差异基因大都是上调表达,主要包括氧化应激蛋白、脱落酸受体、乙烯响应转录因子等。这些差异表达基因可能是西瓜多倍体形成的关键因子。
总之,通过转录组初步分析,我们较为全面地了解了西瓜二倍体与四倍体的转录水平变化,发现了一些与倍性相关的基因,进一步深入研究这些基因的生理功能,有助于揭示多倍体形成的分子机制,丰富人类对植物多倍性的认知。
参考文献:
[1]中华人民共和国农业部. 中国农业统计资料2016[M]. 北京:中国农业出版社,2017.
[2]姚 娜,刘秀明,董园园,等. 转录组的测序方法及应用研究概述[J]. 北方园艺,2017(12):192-198.
[3]Wechter W P,Levi A,Harris K R,et al. Gene expression in developing watermelon fruit[J]. BMC Genomics,2008,9:275.
[4]Guo S G,Liu J A,Zheng Y,et al. Characterization of transcriptome dynamics during watermelon fruit development-sequencing,assembly,annotation and gene expression profiles[J]. BMC Genomics,2011,12(1):454.
[5]Zhu Q L,Gao P,Liu S,et al. Comparative transcriptome analysis of two contrasting watermelon genotypes during fruit development and ripening[J]. BMC Genomics,2017,18:3.
[6]龍娅丽,江雪飞,周 鹏,等. 二倍体西瓜及其同源四倍体叶片sRNA表达谱分析[J]. 热带作物学报,2018,39(4):661-668.
[7]王 涛,陈孟龙,刘 玲,等. 植物多倍体化中基因组和基因表达的变化[J]. 植物学报,2015,50(4):504-515.
[8]Guo S G,Zhang J G,Sun H H,et al . The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions[J]. Nat Genet,2013,45:51-58.
[9]Anders S,Huber W. Differential expression analysis for sequence count data[J]. Genome Biol,2010,11:1-12.
[10]Liu Y,Wei H B,Ma M D,et al. Arabidopsis FHY3 and FAR1 regulate the balance between growth and defense responses under shade conditions[J]. The Plant Cell,2019,31(9):2089-2106.
[11]Young Matthew D,Wakefield M J,Smyth G K,et al. Gene ontology analysis for RNA-seq:accounting for selection bias[J]. Genome Biology,2010,11:R14.
[12]张少平,洪建基,邱珊莲,等. 紫背天葵高通量转录组测序分析[J]. 园艺学报,2016,43(5):935-946.
[13]Kanehisa M,Arak M,Goto S,et al. KEGG for linking genomes to life and the environment[J]. Nucleic Acids Research,2008,36:480-484.
[14]Compton M E,Barnett N,Gray D J. Use of fluorescein diacetate (FDA) to determimne ploidy of in vitro watermelon shoots[J]. Plant Cell,Tissue and Organ Culture,1999,58:199-203.
[15]蔡 力. 二倍体和四倍体紫锥菊中叶形态结构及其光合效率的比较研究[D]. 广东:华南农业大学,2016:35.
[16]王家利,王 芳,郭小丽,等. 同源多倍体化效应研究进展[J]. 中国农学通报,2013,29(12):22-29.
[17]施先锋,彭金光,李煜华,等. 西瓜多倍体鉴定方法的研究[J]. 浙江农业科学,2010(2):273-274.陶亚军,尹建国,樊继伟,等. 苏北地区稻麦周年生产力品种组合筛选[J]. 江苏农业科学,2020,48(24):60-66.
猜你喜欢
今日农业(2020年14期)2020-12-14
诊断学(理论与实践)(2020年6期)2020-12-09
临床与实验病理学杂志(2017年3期)2017-03-07
文理导航·趣味课堂(2016年6期)2016-09-09
中学生数理化·高考版(2008年12期)2008-06-17
