
PVA微晶交联和SA/PAA双网络协效增强增韧SA纤维
2020-02-19 07:32周炜东石军峰赵云鹤叶泳铭
高等学校化学学报 2020年2期
闫 铭, 周炜东, 张 鸿, 石军峰, 赵云鹤,叶泳铭, 郭 静, 于 跃
(大连工业大学纺织与材料工程学院, 大连 116024)

近年来, 科研人员通过将SA水凝胶与其它物质复合形成双网络结构[18,19]对SA水凝胶进行改性. 龚剑萍等[20]首次以聚2-丙烯酰胺基-2-甲基丙磺酸(PAMPS)和聚丙烯酰胺(PAAm)为原料制备了双网络水凝胶. 水凝胶是极好的软物质材料, 可以看作大分子网络对水溶液的增强, 并赋予其一定的形状和机械强度. 因为双网络结构能够克服传统水凝胶内部化学交联点分布不均及力学性能差的缺陷, 所以广泛应用于水凝胶领域. 孙天文等[21]成功制备了丙烯酰胺/海藻酸钠双网络水凝胶, 研究结果表明, 双网络水凝胶的力学性能更加稳定, 溶胀率更稳定且快速达到溶胀平衡. Bahrami等[22]在SA和N,N-亚甲基双丙烯酰胺(MBA)存在下, 经自由基聚合获得PAAm第一个化学交联网络, 再将获得的SA/PAAm水凝胶浸入八苯基溶液中形成第二物理交联网络, 进而制备混合水凝胶, 研究结果表明, 混合水凝胶的抗压强度和机械性能显著提高. 杨曼丽等[23]先制备了海藻酸钠/聚丙烯酸(SA/PAA)凝胶, 再添加纳米二氧化硅颗粒增强增韧, 制得了海藻酸钠/聚丙烯酸/纳米二氧化硅复合膜, 研究结果表明, 复合膜的最大抗张强度和最高断裂伸长率与纯海藻酸钠相比分别提高了126.7%和97.1%. 当前, 双网络结构主要应用于水凝胶及膜等领域, 很少应用于纤维领域, 这是因为利用双网络制备纤维时, 第二网络的引入通常会增加溶液的黏度, 而湿法纺丝时要求溶液黏度不能过高, 这就导致很难将双网络用于纤维改性.
Zhu等[24]将SA与丝素蛋白(SF)复合以提高SA纤维的力学性能, 并且可以采用双网络进行结构设计和增韧改性. 本文在低温溶解和湿法纺丝基础上, 设计了SA/PAA双网络结构和聚乙烯醇(PVA)微晶交联结构协效改性SA纤维, 研究了纺丝原液的流变性能和复合纤维的结构与性能.
1 实验部分
1.1 试剂与仪器
SA, 食品级, 青岛明月海藻集团有限公司; 氢氧化锂(LiOH)、 尿素(Urea)、 过硫酸铵(APS)、 丙烯酸(AA)、 聚乙烯醇(PVA)和MBA, 分析纯, 上海麦克林生化科技有限公司.
CP214型电子天平, 奥豪斯仪器(上海)有限公司; DHJF Series型低温恒温水浴锅, 郑州长城科工贸有限公司; TDL-80-2B型离心机, 济南来宝科学仪器有限公司; KQ2200B型超声波清洗器, 昆山市超声仪器有限公司; DNG-800S型变频步进器, 纽格拉思公司; DHR-2型旋转流变仪, 美国TA公司; LL-06E型电子单纤强力测试仪, 莱州市电子仪器有限公司; Spectrum two型傅里叶变换红外光谱(FTIR)仪, 美国铂金埃尔默公司; D/max-3B型X射线衍射(XRD)仪, 日本理学公司; JSM-6460LV型扫描电子显微镜(SEM), 日本电子公司.
1.2 实验过程
1.2.1 低温溶解制备SA/LiOH/Urea溶液 将5.5 g LiOH置于烧杯中, 加入100 mL去离子水, 再加入质量分数为20%的Urea配制成溶液, 用AA调节溶液pH值至10~10.5, 加入SA后, 将上述溶液于-12 ℃放置12 h, 继续采用冷冻-融解方法溶解SA, 每次冷冻循环取样融解真空脱泡得到纺丝液; 将剩下的溶液再放入冰箱中冷冻, 共循环4次.
1.2.2 再生SA纤维的制备 将SA/LiOH/Urea纺丝液经变频步进器以9 mm/min速率从孔径为0.7 mm的喷丝孔挤出, 进入质量分数为3%的氯化钙(CaCl2)的溶液中凝固成型, 经水洗、 牵伸及干燥得到再生SA纤维.
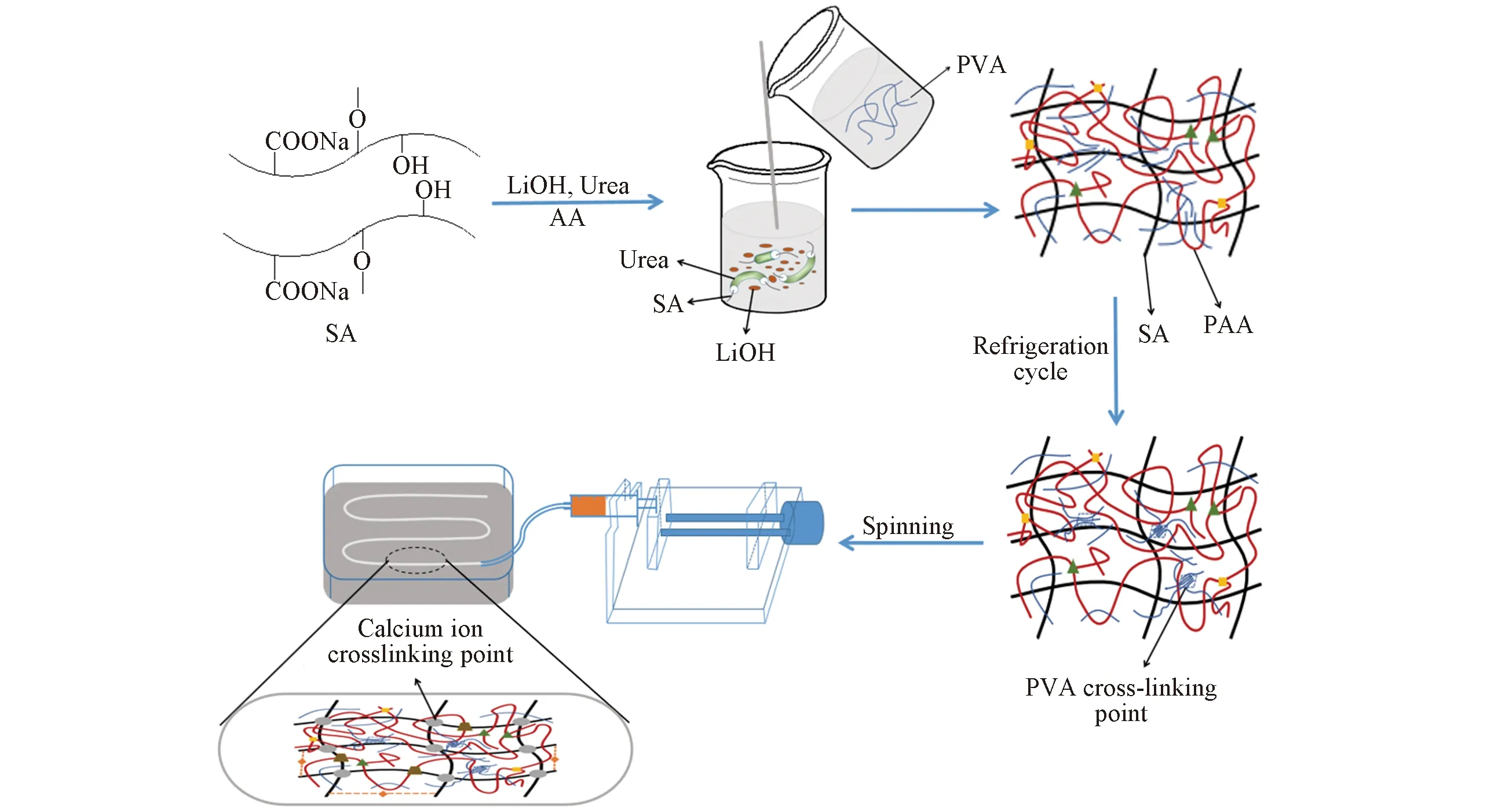
Scheme 1 Preparation of flow chart
1.2.3 SA/PAA/PVA复合纤维的制备 Scheme 1给出SA/PAA/PVA复合纤维的合成路线. 将一定质量的PVA放入烧杯中, 加入100 mL去离子水, 于90 ℃溶解2 h后静置, 得到PVA溶液; 用AA调节SA/LiOH/Urea溶液的pH值至6~6.5, 依次在溶液中分别加入0, 0.25%, 0.5%, 0.75%和1%的MBA和0.5%的APS(其中MBA和APS的含量均为占AA的质量分数); 于80 ℃水浴加热2 h, 冷却到室温后, 将得到的溶液与PVA溶液按体积比10∶3混合, 再于40 ℃恒温加热1 h, 得到纺丝液; 将纺丝液以9 mm/min速率从孔径为0.7 mm的喷丝孔挤出, 进入3%的CaCl2溶液中凝固成型, 经过水洗、 牵伸及干燥, 得到改性的SA/PAA/PVA复合纤维.
2 结果与讨论
2.1 SA/PAA/PVA共混溶液的流动性能

Fig.1 Characterization of rheological properties of SA/PAA/PVA blend solutiona—g. Loss modulus; a′—g′ storage modulus. a, a′. Regenerated SA; b, b′. SA/PAA/PVA, not frozen; c, c′. SA/PAA/PVA, already frozen; d, d′. SA/PAA/PVA, 0.25%MBA; e, e′. SA/PAA/PVA, 0.5%MBA; f, f′. SA/PAA/PVA, 0.75%MBA; g, g′. SA/PAA/PVA, 1%MBA.
图1给出不同MBA含量的SA/PAA/PVA共混溶液和再生SA的流变测试结果. 由图1可见, 未冷冻的SA/PAA/PVA共混溶液的储能模量(G′)和损耗模量(G″)均大于再生SA溶液, 这是因为加入PVA之后, PVA与SA形成了氢键, 分子间作用力增大, 从而增加共混溶液的弹性和黏性; 冷冻4次后, SA/PAA/PVA共混溶液的G′和G″均增加, 说明经冷冻后的共混溶液的弹性和黏性均增加, 这是因为冷冻后产生了PVA微晶交联点, 防止分子间相对滑移, 从而增加溶液的黏性和弹性. 复合体系临界松弛指数(n)可以利用损耗角(δ)通过公式tanδ=G″/G′=tan(nπ/2)计算, 当n<0.5时,G′>G″, 说明微晶交联结构比较完善[25]. 再生SA溶液、 未冷冻SA/PAA/PVA共混溶液和冷冻后的SA/PAA/PVA共混溶液的n值分别为0.55, 0.52和0.49. 冷冻后的SA/PAA/PVA共混溶液的n值明显低于再生SA和未冷冻SA/PAA/PVA体系, 并且n<0.5, 表明G′>G″, 微晶结构产生且微晶交联结构比较完善.
在未冷冻体系中, 频率相同时, 交联剂MBA含量对SA/PAA/PVA共混溶液的G′和G″有影响. 在MBA含量为0.75%和1%时,G′>G″, 说明其弹性大于黏性, 这是因为交联剂过多, PAA交联度大, 从而SA与PAA网络缠结较多, SA分子链滑移困难. 在MBA含量为0.25%和0.5%时, SA/PAA/PVA共混溶液的G″>G′, 说明其黏性大于弹性, 为假塑性流体. 这是因为, 一方面SA与PAA分子链缠结较少, 在一定程度上还可以发生解缠结, SA分子链可以发生移动, SA与PAA分子间相互作用力不是很强; 另一方面, SA/PAA的双网络结构使SA分子间距离变大, 从而导致SA与SA分子间作用力变小. SA/PAA复合纤维双网络结构形成示意图如Scheme 2所示.

Scheme 2 Schematic diagram of SA/PAA composite fiber dual network structure
由图1可知, MBA含量为0.5%的共混溶液的G″小于MBA含量为0.25%的共混溶液, 这是因为MBA含量为0.25%时, PAA化学交联网络较少, 有一定量的PAA以分子链的形式存在, PAA对SA间分子作用的影响有限; 而当MBA含量为0.5%时, 绝大多数PAA形成了化学交联网络, 但因交联程度较小, 对SA链段运动并无明显束缚, 而且PAA网络的存在使得SA分子间距增大, 分子间作用力减少, 所以溶液黏性降低. 因此当MBA含量为0.5%时, SA/PAA/PVA共混溶液的流动性能更好, 为纤维成型提供了良好的可纺性.
2.2 SA/PAA/PVA复合纤维的力学性能

Fig.2 Breaking strength(a) and breaking elongation(b) of SA/PAA/PVA composite fibers at different MBA contents
图2给出交联剂含量对SA/PAA/PVA复合纤维的断裂强度和断裂伸长率的影响. 由图2可见, 随着MBA含量的增加, SA/PAA/PVA复合纤维的断裂强度和断裂伸长率先增加后减小, 当MBA的含量为0.5%时, 复合纤维的断裂强度和断裂伸长率出现最大值, 断裂强度达到2.83 cN/dtex, 断裂伸长率达到9.38%. 一方面, 在MBA含量为0.5%时, SA与PAA形成双网络结构, PAA柔性网络的存在可以使SA分子间距离变大, 导致分子间作用力减小, 从而提高SA刚性大分子链的移动能力; 另一方面, 即使部分Ca2+离子交联结构受到损害, PAA柔性网络仍可以利用其弹性来保持SA/PAA/PVA复合纤维的整体结构不被破坏, 所以在拉伸过程中双网络互锁结构可以均化网络, 并且在低温下PVA形成微晶, 可以成为新的物理交联点[26], 可以协同耗散复合纤维的负载能量, 从而提高复合纤维的断裂强度和伸长率; 第三方面原因是, SA和SA, SA和PAA, PAA和PAA之间存在大量动态牺牲键(Scheme 3), 可在受力时断裂并重组耗散能量来抑制应力集中现象的发生, 增加复合纤维的断裂强度和断裂伸长率.
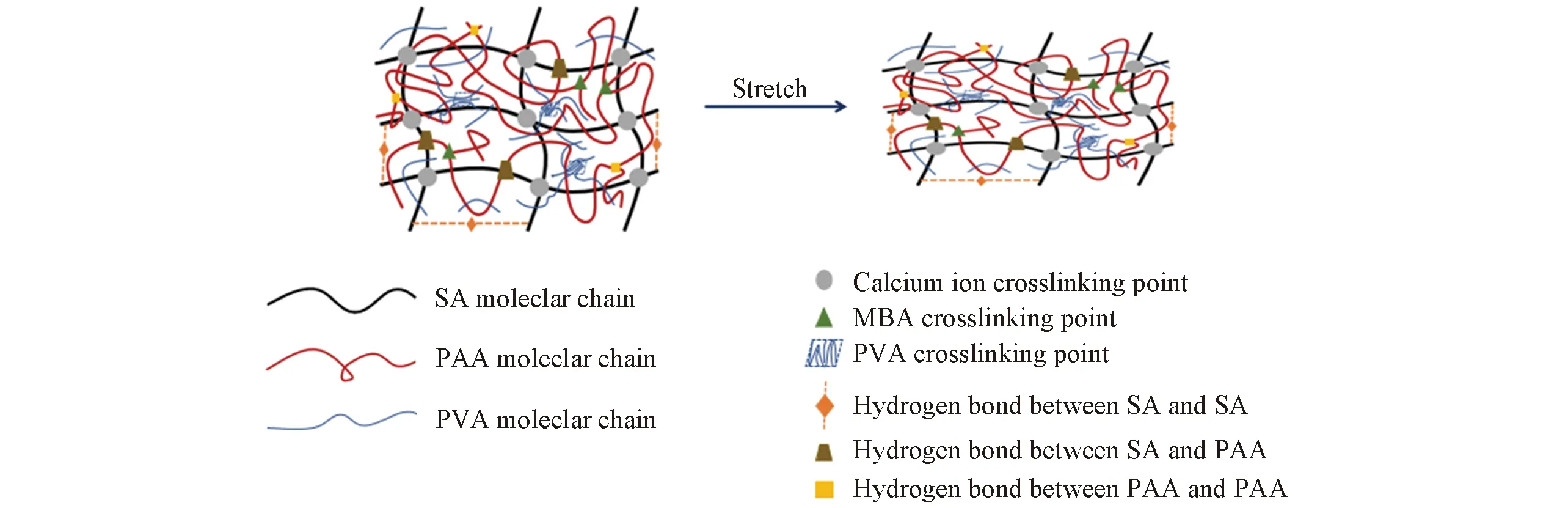
Scheme 3 Roles of dynamic victim keys
图3给出冷冻处理对SA/PAA/PVA复合纤维力学性能的影响. 由图3可见, 断裂强度的相对大小依次为: 含0.5%MBA冷冻SA/PAA/PVA复合纤维>冷冻后SA/PAA/PVA复合纤维>未冷冻SA/PAA/PVA复合纤维>再生SA纤维. 再生纤维中加入PVA后, PVA与SA形成氢键, 分子间作用力增大, 所以未冷冻处理时, SA/PAA/PVA复合纤维的力学性能已有提高; 冷冻后SA/PAA/PVA复合纤维的力学性能提高是因为PVA微晶的产生增加了微晶物理交联点承受并耗散外力[26]; 含0.5%MBA的冷冻SA/PAA/PVA复合纤维的力学性能进一步提高主要是因为形成了含PVA柔性链结构的SA/PAA双网络互锁结构.
由SA/PAA/PVA共混溶液的流变测试和SA/PAA/PVA复合纤维的力学性能测试可知, 经过4次冷冻-融解循环处理并且当MBA含量为0.5%时溶液的流动性能最佳, 力学性能也最好, 故以此条件为最佳测试条件.
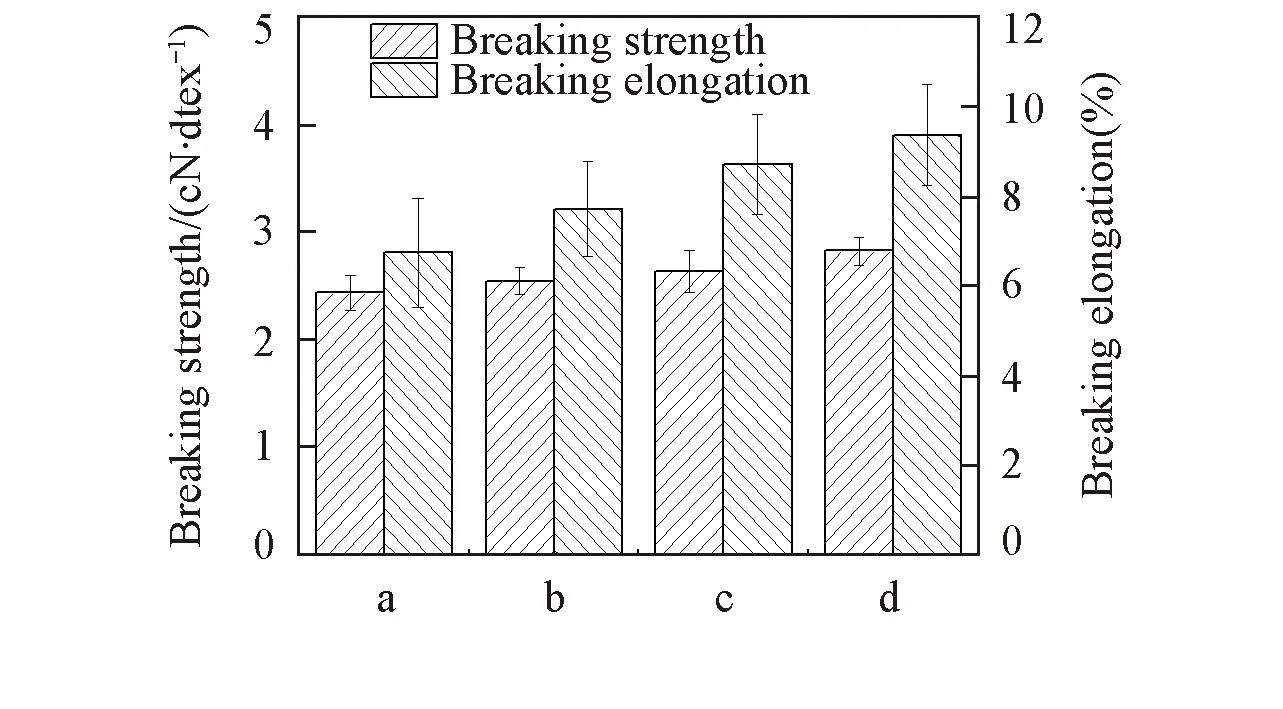
Fig.3 Mechanical properties of regenerated SA fiber and SA/PAA/PVA composite fibers treated under different conditionsa. Regenerated SA; b. not frozen, SA/PAA/PVA; c. 0.25% MB and already frozen, SA/PAA/PVA; d. 0.5% MBA and already frozen, SA/PAA/PVA.
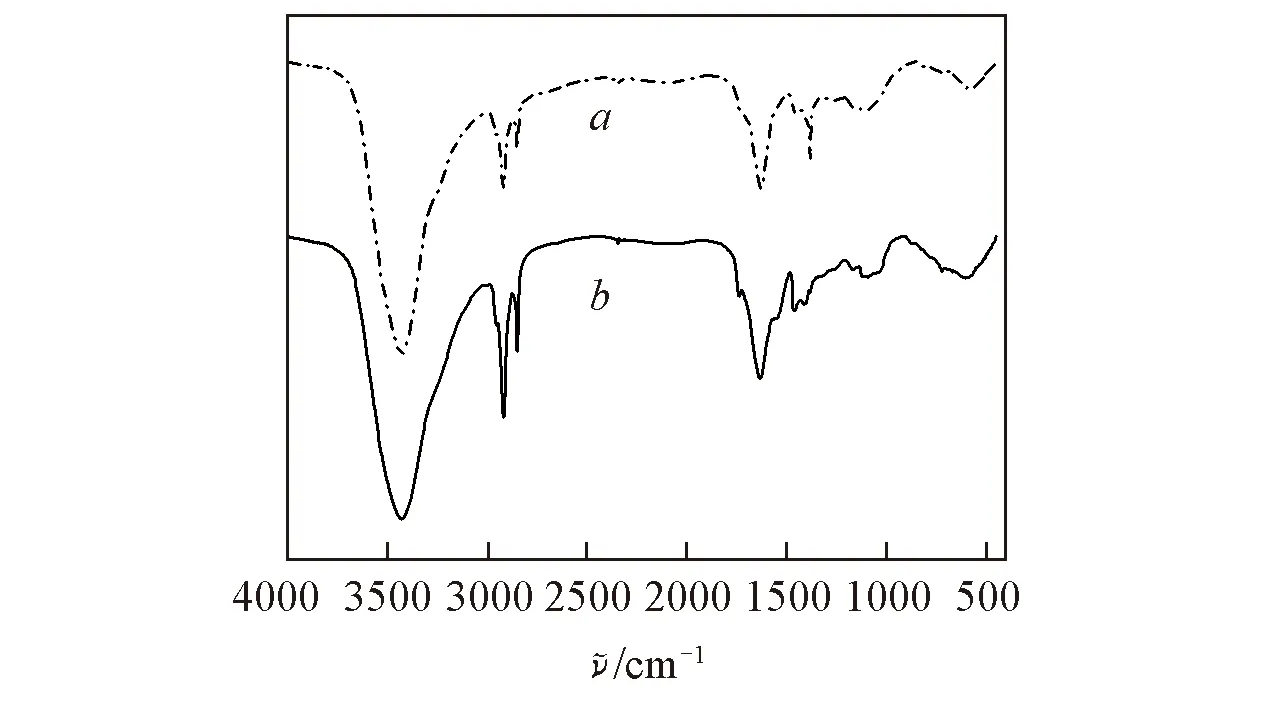
Fig.4 Infrared spectra of regenerated SA fiber(a) and SA/PAA/PVA composite fiber(b)
2.3 SA/PAA/PVA复合纤维的化学结构

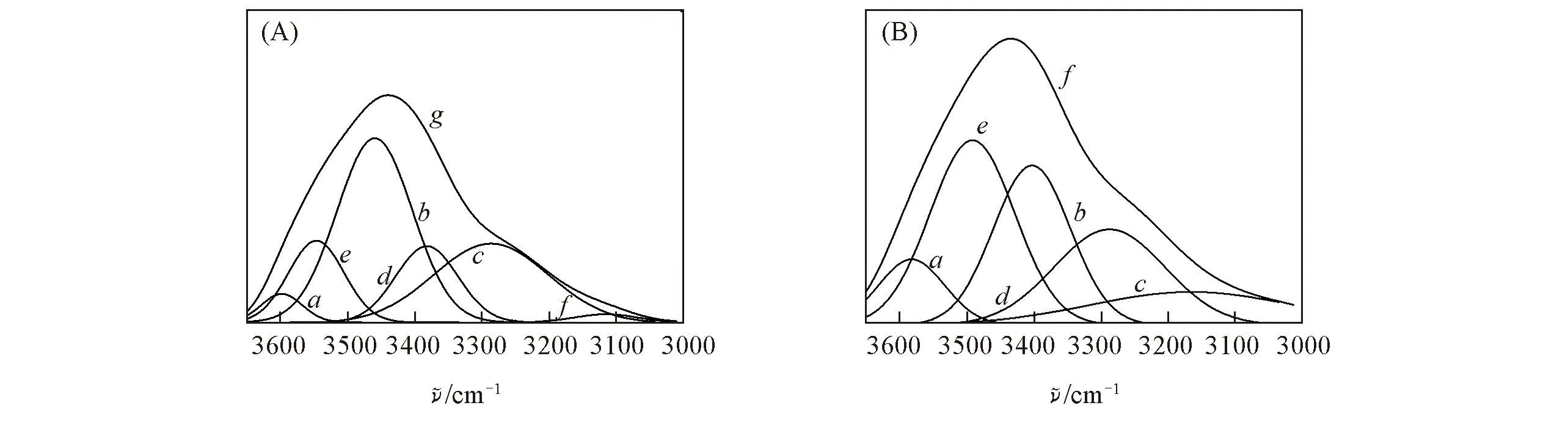
Fig.5 Fitted IR curves of regenerated SA fiber(A) and SA/PAA/PVA composite fiber(B) (A) a—f. Fitted curves 1—6; g. originaL curve. (B) a—e. Fitted curves 1—5; f. original curve.
对再生SA纤维和SA/PAA/PVA 复合纤维在3000~3650 cm-1(环氧—CH2峰)范围内的红外谱图进行分峰处理. 图5给出再生SA纤维和SA/PAA/PVA复合纤维羟基峰处的高斯峰拟合及其子峰分布(表1). 由表1可见, 与再生SA纤维相比, 复合纤维自由羟基所占的比例由3.5%上升到8.9%, 分子内氢键所占的比例由54.2%下降到37.1%, 分子间氢键所占的比例由42.3%上升到54.0%, 说明将PAA和PVA引入体系后, 由于含有大量的羟基, 羟基先与剩下的醚O键结合再与π键结合, SA/PAA/PVA复合纤维分子间氢键比例呈增加趋势, 与上述红外测试中—OH峰变宽并且红移的结果一致. 分子间氢键增加后, 会相应地减少分子内氢键形成的结合位点, 导致分子内氢键比例下降. 羟基结合成分子内氢键和分子间氢键后还会有所剩余, 因此自由羟基的比例也会增加, 即外加的—OH与SA上的醚O键、π键和羟基的结合速度顺序为—OH与醚键>—OH与π键>—OH与羟基.
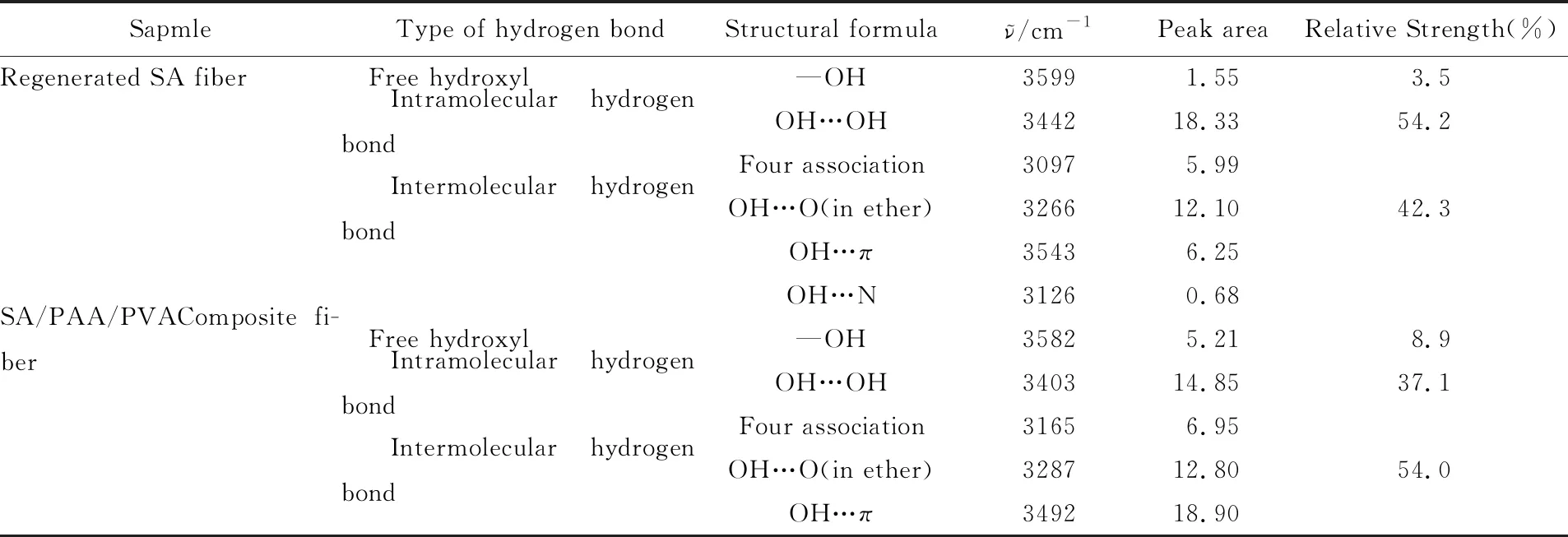
Table 1 Fractal peak fitting results of infrared spectrum of regenerated SA fiber and SA/PAA/PVA composite fiber
2.4 SA/PAA/PVA复合纤维的结晶性能
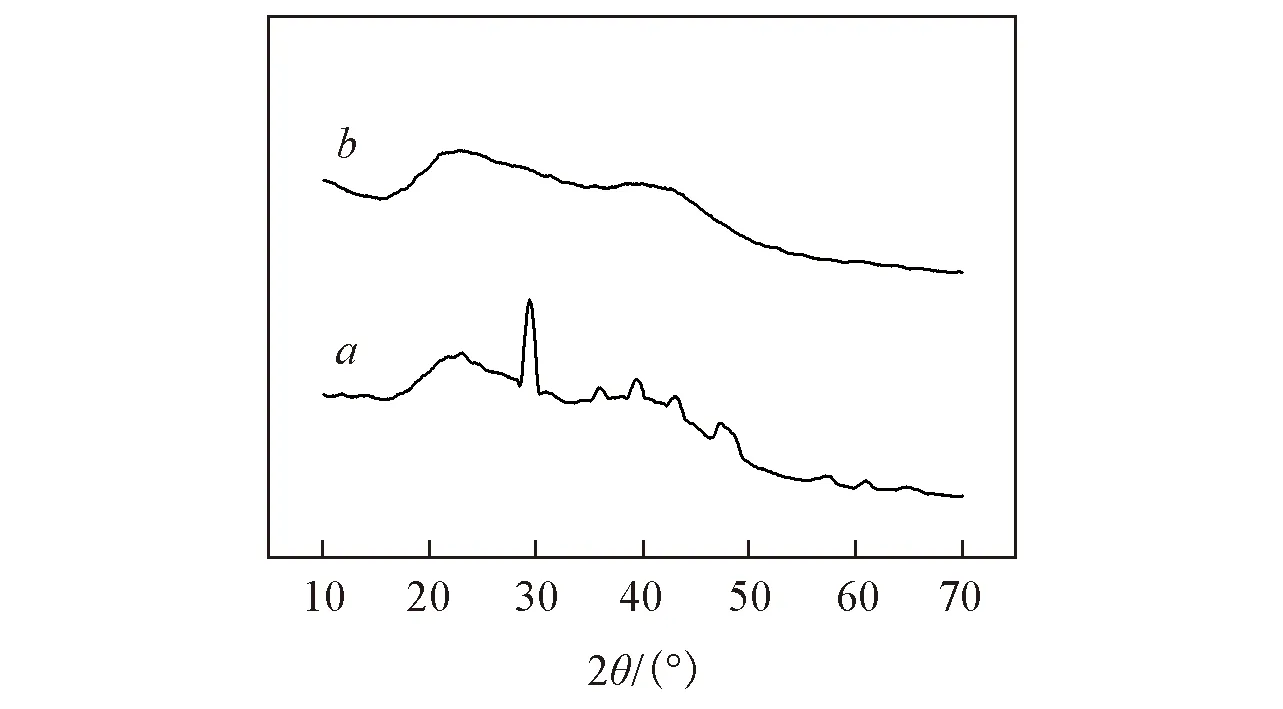
Fig.6 XRD patterns of regenerated SA fiber(a) and SA/PAA/PVA composite fiber(b)
图6给出SA/PAA/PVA复合纤维与再生SA纤维的XRD谱图. 由图6可知, 在2θ=19.9°时出现PVA的特征峰, 在2θ=22°和41°时, SA/PAA/PVA复合纤维峰的强度比再生SA的峰强度高, 表明复合纤维的结晶性能比再生SA的结晶性能好. SA/PAA/PVA复合纤维和再生SA纤维的结晶度分别为32.47%和6.03%, PAA和PVA的加入提高了复合纤维的结晶度是因为PAA和PVA引入到体系后, SA与PAA, SA与PVA, PAA与PVA之间均含有羟基与羟基的相互作用而形成氢键结构, 同时PVA柔性链易于流动取向诱导结晶, 并且冷冻处理促进了PVA微晶结构生成, 从而提高了复合纤维的结晶度.
2.5 SA/PAA/PVA复合纤维的热稳定性
图7给出SA/PAA/PVA复合纤维与再生SA纤维的TG及DTG曲线. 可以看出, SA/PAA/PVA复合纤维和再生SA纤维分解的3个阶段对应的温度区间相似: 第一阶段对应温度区间30~200 ℃, 主要是SA内部自由水和结合水的扩散损失. 第二阶段对应温度区间200~390 ℃, 主要是SA大分子结构单元间糖苷键的断裂和SA大分子内部和大分子链间临近羟基以水分子形式脱去, 此时SA大分子骨架开
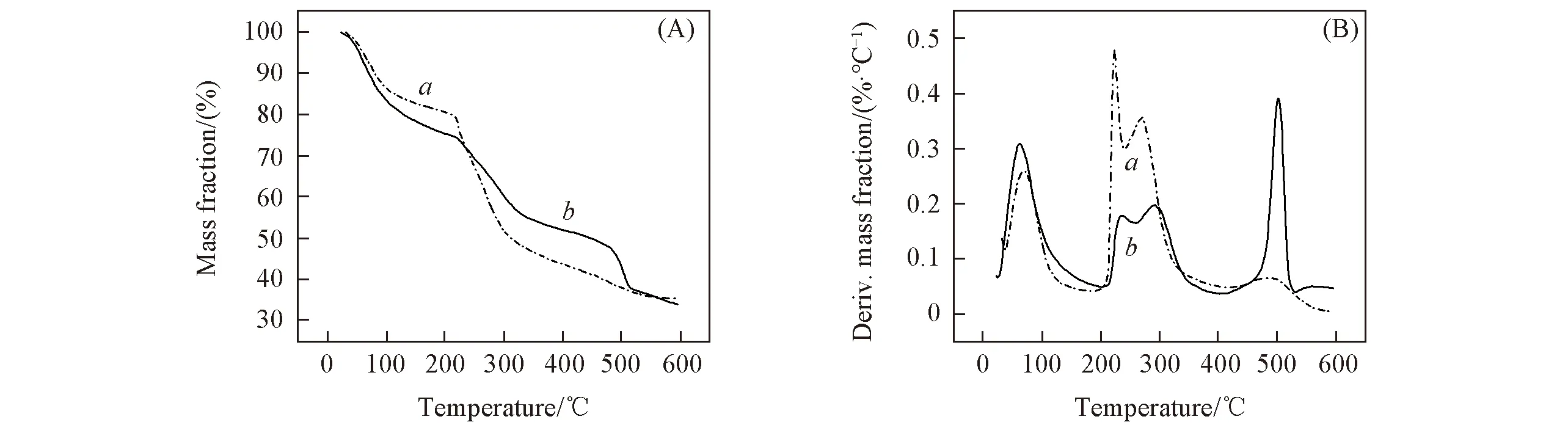
Fig.7 TG(A) and DTG(B) curves of regenerated SA fiber(a) and SA/PAA/PVA composite fiber(b)
始断裂, SA在该阶段炭化分解为中间产物, 但在该阶段, SA/PAA/PVA复合纤维的失重率明显低于再生SA纤维, 这是因为相比于具有单一离子交联网络结构的SA纤维, 一方面SA/PAA双网络结构中的PAA化学交联柔性网络与SA大分子离子交联网络互锁, 另一方面SA与PAA具有很强的分子间作用力, 从而SA/PAA/PVA复合纤维的热稳定性能增强; PVA在冷冻循环过程中形成的微晶交联点也能提高纤维的热稳定性能; SA/PAA形成的双网络结构, 在燃烧时有利于形成连续的炭层, 降低复合材料的燃烧速度, 延缓燃烧进程, 提升复合材料的热稳定性[30]. 第三阶段对应温度区间410~550 ℃, 该区间对应SA形成的中间产物进一步分解. 在该阶段, SA/PAA/PVA复合纤维的失重率明显高于再生SA纤维, 这是因为PVA在450 ℃时发生分子链的无规则断链[31]并且PAA在500 ℃时开始分解[32].
2.6 SA/PAA/PVA复合纤维的形貌分析
图8给出SA/PAA/PVA复合纤维和再生SA纤维的表面及断面SEM照片. 由图8(A)和(B)可见, SA/PAA/PVA复合纤维的表面比再生SA纤维表面更规整、 均匀、 光滑, 表面原纤化结构减少, 沟槽数量减少. 这是因为PAA和PVA加入到体系后, 分别与SA产生相互作用力, 导致SA与SA大分子链间氢键作用被减弱, 继而SA的分子间距有所增加, 所以Ca2+更容易均匀地扩散到SA分子内部, 在双扩散过程中, 复合纤维更加趋向均匀收缩. 由图8(C)和(D)可见, SA/PAA/PVA复合纤维的断面比再生SA纤维断面更致密, 并且愈发聚集, 这是因为再生SA纤维还存在一定的相分离, 而加入PAA和PVA后的SA/PAA/PVA复合纤维发生强迫互容, 宏观上表现为复合纤维的断面更加致密.

Fig.8 SEM images of surface(A, B) and cross-sectional(C, D) of regenerated SA fiber(A, C) and SA/PAA/PVA composite fiber(B, D)
3 结 论
通过向体系内加入AA和PVA, 利用湿法纺丝和冷冻处理的方法可形成PVA微晶交联和SA/PAA双网络结构对SA纤维进行协效改性. 外加的—OH与SA上的醚O键结合的速度>与π键结合的速度>与羟基结合的速度; 当MBA含量为0.5%时, 复合纤维的断裂强度达到2.83 cN/dtex, 断裂伸长率达到9.38%, 比再生SA纤维分别提高了15.98%和38.96%; PAA和PVA已经成功复合到体系中; PAA和PVA的加入提高了复合纤维的结晶度, 纤维的表面形貌趋于光滑、 规整, 纤维断面更加致密.
猜你喜欢
高中数理化(2022年14期)2022-08-15
陶瓷学报(2021年4期)2021-10-14
建材发展导向(2021年11期)2021-07-28
陶瓷学报(2021年1期)2021-04-13
原子与分子物理学报(2019年5期)2019-04-28
制造技术与机床(2017年4期)2017-06-22
系统工程与电子技术(2016年7期)2016-08-21
系统工程与电子技术(2016年7期)2016-08-21
北京信息科技大学学报(自然科学版)(2016年6期)2016-02-27
中学化学(2015年12期)2016-01-19

- 高等学校化学学报的其它文章
- 可双光子激发的聚集诱导发光光敏剂及其生物医学应用
- 金属有机框架材料的结构、 动力学行为和主客体相互作用的固体核磁共振研究
- 大面积多元化表面等离激元金纳米粒子结构的制备
- Determination of Triazine Herbicides from Fruit Juice Samples Using Effervescence Assisted Microextraction Method Based on Acidic Ionic Liquid Packed Syringe
- 聚谷氨酸接枝聚乙二醇@碳酸钙遮蔽体系用于提高聚乙烯亚胺基因转染效率
- 基于萘普生-芳基金属配合物的抗癌及抗炎性能