
原位溶剂热聚合法制备金属有机骨架/碳化氮纳米片涂覆的固相微萃取纤维用于红茶中农药残留的高灵敏检测
2020-02-19 09:05张文敏冯遵梅黄川辉
色谱 2020年3期
张文敏, 冯遵梅, 黄川辉, 高 佳, 张 兰,*
(1. 闽江师范高等专科学校, 福建 福州 350109; 2. 福州大学, 食品安全与生物分析教育部重点实验室, 福建 福州 350116)
有机氯(organochlorine pesticides, OCPs)和拟除虫菊酯(pyrethroids, PYs)是两类广泛用于控制植物病虫害的农药。由于OCPs和PYs生物降解性低、持久性高,会严重污染自然环境[1,2]。此外,有研究表明OCPs对人类身体有潜在的毒性,例如损伤人类神经系统、破坏细胞免疫系统和造成生殖障碍等[3,4]。因此,OCPs已被列为持久性有机污染物并被联合国环境规划署禁止使用。茶叶是中国流行的传统饮品,对人的身体健康有着积极的影响。由于其独特的香气、风味和益处,茶叶在中国乃至世界各地都广受欢迎。欧盟和日本等对茶叶中农药残留量的最大限量制定了严格标准,尤其是对于PYs和OCPs。因此,为了减少对自然环境的污染、保障民众的食品安全和促进国际茶叶贸易,急需建立一种高效、灵敏的分析方法用于茶叶中PYs和OCPs的检测[2]。
目前,高效液相色谱(HPLC)[5-8]和气相色谱(GC)[9-17]是最常用的检测农药残留的分析方法。然而,由于OCPs和PYs的含量相对较低以及样品基质的复杂性,在进行色谱分析之前,有效的预处理和富集过程是必不可少的。其中,固相微萃取(solid-phase microextraction, SPME)因其简单、灵敏且不需要溶剂解吸等优点,被广泛应用于样品前处理过程[9-13]。在SPME方法中,纤维涂层材料又与SPME的萃取效率息息相关,是影响其灵敏度、选择性、稳定性和工作寿命的关键因素。
金属有机骨架材料(metal organic frameworks, MOFs)的出现为科学家打开了设计纳米多孔材料的新世界。MOFs是由金属离子或金属簇和有机配体连接组成的开放式多孔三维结构,具有高比表面积、结构可调性、高孔隙率以及可修饰性等特性,是SPME方法中最有魅力的涂层材料之一[18]。但是,大多数MOFs在水溶液和弱酸性溶液中稳定性差,晶体结构易被破坏,限制其应用范围。近年来,制备MOFs复合纳米材料成为提高MOFs稳定性和丰富其与目标物相互作用力的有效途径之一。Li等[19]通过一种简单的多层粒子间连接方法制备稳定且厚度可控的MOF-199/CNTs涂层材料,并成功地应用于葡萄、黄皮、蓝莓和榴莲等相对潮湿水果样品中的痕量乙烯的无损分析。
多孔碳化氮纳米片(HOCN)是一种新兴的二维层状纳米材料,具有丰富的活性位点和高度可接近的表面,对目标物具有优异的萃取性能,已成功用于SPME方法中[9]。在本研究中,尝试将经典的MOFs纳米材料UiO-66与HOCN纳米片相结合,通过原位溶剂热聚合法制备UiO-66/HOCN复合材料为涂层的SPME纤维,对其稳定性以及对9种使用广泛的农药的萃取效果进行考察。将其与GC-MS相结合,经过实验条件的优化,建立了一种适用于OCPs和PYs痕量检测的分析方法,并考察了该方法的检出限、回收率和重现性等。最后,将所建立的分析方法用于实际红茶样品中OCPs和PYs的检测。
1 实验部分
1.1 仪器与试剂
TRACE1300/TSQ 8000 Evo气相色谱-质谱联用仪(美国Thermo Fisher Scientific公司); Tecnai G2 F20透射电子显微镜(美国FEI公司); JSM-6300F扫描电子显微镜(日本JEOL公司); EscaLab 250Xi X-射线光电子能谱仪(美国Thermo Fisher Scientific公司); Nicolet 6700傅里叶变换红外光谱仪(美国Thermo Fisher Scientific公司); ASAP 2020物理吸附仪(美国Micromeritics公司); DIL402C热重分析仪(德国Netzsch公司); Nano 2S纳米粒度Zeta电位分析仪(英国Malvern公司); ZBCL-GS智能数显磁力搅拌加热锅(予华仪器科技有限公司)。
9种农药标准品(1.0 g/L)购自中国Aladdin试剂有限公司,分别为七氯(heptachlor)、艾氏剂(Aldrin)、环氧七氯(heptachlor epoxide)、α-氯丹(α-chlordane)、p,p′-滴滴伊(p,p′-DDE)、α-硫丹(α-endosulfan)、p,p′-滴滴滴(p,p′-DDD)、p,p′-滴滴涕(p,p′-DDT)及联苯菊酯(bifenthrin);对苯二甲酸(p-phthalic acid, PTA,纯度99%)、氯化锆(ZrCl4,纯度99.9%)和3-氨丙基三乙氧基硅烷((3-aminopropyl)triethoxysilane, APTES,纯度98%)购自中国Aladdin试剂有限公司;N,N-二甲基甲酰胺(N,N-dimethylformamide, DMF,纯度99.5%)、甲醇(methanol,纯度99.5%)、丙酮(acetone,纯度99.5%)、氯化钠(NaCl,纯度99.5%)和氨水(25%~28%,质量分数)购自中国国药试剂有限公司。实验用水均为Milli-Q净水器所制得的超纯水(18.2 MΩ·cm)。
1.2 标准溶液的配制
七氯、艾氏剂、环氧七氯、α-氯丹、p,p′-滴滴伊、α-硫丹、p,p′-滴滴滴、p,p′-滴滴涕及联苯菊酯标准溶液(1.0 g/L)储备液于4 ℃下避光保存。系列标准溶液用丙酮配制,现配现用。
1.3 UiO-66/HOCN复合材料的制备
1.3.1HOCN纳米片的制备
根据文献[20]报道的方法合成HOCN黑色粉末。将获得的100.0 mg HOCN粉末加入到25.0 mL HCl(10 mol/L)溶液中,磁力搅拌处理1 h后,以5 000 r/min离心10 min收集沉淀物,并用超纯水洗涤干净。将所获得的沉淀物重新分散在100.0 mL超纯水中,超声处理6 h以形成稳定的HOCN纳米片分散溶液。最后,将所获得的HOCN纳米片分散溶液冷冻干燥,以用于UiO-66/HOCN复合材料的制备。
1.3.2UiO-66/HOCN复合材料的制备
UiO-66/HOCN复合材料通过原位水热法制备,用于该复合材料的一些性能表征。具体过程如下:分别将82.0 mg的ZrCl4溶于10.0 mL的DMF溶液中作为A液;将58.0 mg PTA溶于30.0 mL的DMF溶液中作为B液;将29.0 mg HOCN均匀分散在30.0 mL的DMF溶液中作为C液。将A液加入到B液中,磁力搅拌5 min后,再将上述混合溶液加入到C液中,继续磁力搅拌5 min。将所获得的混合溶液加入到100 mL高压反应釜中,在120 ℃下反应48 h后,自然冷却至室温,将所获得的产物用DMF和甲醇交替洗涤3次(8 000 r/min, 5 min),并在60 ℃下真空干燥,以获得UiO-66/HOCN复合材料。在UiO-66/HOCN复合材料优化实验中,保持其他制备条件不变,通过改变HOCN占PTA的质量分数(0~100%),分别表示为UiO-66/HOCN-n(n=0, 10, 30, 50, 70, 90和100)复合材料。
1.4 SPME纤维的制备
由于不锈钢纤维具有化学惰性,通过银镜反应将不锈钢纤维镀上银层之后,利用银和氨基的配位作用使不锈钢纤维易于化学修饰。具体步骤如下:将不锈钢纤维的一端(4.0 cm)浸入氢氟酸溶液中30 min后,用超纯水清洗,以获得一定直径的粗糙表面。之后将不锈钢纤维的蚀刻部分浸入10.0 mL的AgNO3溶液(0.2 mol/L)中,在剧烈搅拌下,滴加氨水(25%~28%,质量分数)到上述溶液中,有棕色沉淀产生,继续向溶液中滴加氨水(2.8%,质量分数)直至沉淀消失。将获得的[Ag(NH3)]+溶液连同不锈钢纤维快速转移至1.0 mol/L(10.0 mL)葡萄糖溶液中静置约1 h以获得Ag层。再将Ag涂覆的纤维快速插入5.0 mL的APTES溶液中静置12 h后,放入60 ℃烘箱中以完成硅烷化反应。最后将官能化纤维放入含有ZrCl4、PTA和HOCN混合溶液的高压反应釜中(ZrCl4、PTA和HOCN混合溶液的制备步骤请参照1.3.2章节),在120 ℃下反应48 h后,以获得UiO-66/HOCN复合材料涂覆的SPME纤维。
1.5 气相色谱和质谱条件
1.5.1气相色谱条件
TG-5 MS石英毛细管柱(30 m×0.25 mm×0.25 μm);载气:高纯He(纯度>99.999%);流速:恒流,1.5 mL/min;进样方式:无分流进样;进样口温度:280 ℃;升温程序:40 ℃保持1.5 min,以25 ℃/min升温至90 ℃保持1.5 min,以25 ℃/min升温至180 ℃,以5 ℃/min升温至280 ℃,以10 ℃/min升温至300 ℃保持6 min,总运行时长为36.6 min。
1.5.2质谱条件
离子源:EI源;电离能量:70 eV;溶剂延迟:10 min;四极杆温度:150 ℃;接口温度:300 ℃;离子源温度:300 ℃;萃取条件优化部分数据的采集方式:全扫描模式(Scan);质量扫描范围(m/z): 50~550;定量部分的数据采用选择离子监测模式(SIM)进行采集(见表1)。

表 1 9种农药的保留时间、定量离子及定性离子Table 1 Retention times, quantitative ions and qualitative ions for the nine pesticides
1.6 样品的制备
茶叶样品来源于福建省茶叶质量检测中心。样品制备过程参照国家食品安全标准(GB/T 23204-2008)并略有改进。称取5.0 g试样于50 mL离心管中,加入15.0 mL乙腈,超声提取30 min, 5 000 r/min离心3 min,吸取上清液于100 mL圆底烧瓶中。残渣用20.0 mL乙腈重复提取一次,合并全部提取液。然后将所获得的提取液在40 ℃下旋转蒸发至近干,用2.0 mL丙酮溶解,在4 ℃冰箱中储存备用。取20.0 μL的实际样品溶液用丙酮配制成20.0 mL的待测液用于实际样检测。
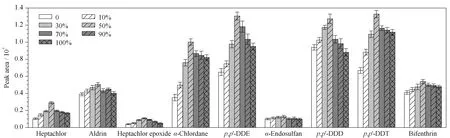
图 1 UiO-66/HOCN复合材料中不同HOCN掺杂比例对9种农药的萃取性能(n=3)Fig. 1 Extraction performance of UiO-66/HOCN composite materials with various contents of HOCN for the nine pesticides (n=3) Experimental conditions: pesticide concentration, 100.0 ng/L; extraction temperature, 50 ℃; extraction time, 40 min; stirring rate, 500 r/min; desorption temperature, 280 ℃; desorption time, 5 min.
1.7 SPME过程
将萃取纤维插入含有20.0 mL样品或标准品溶液的玻璃瓶中,并将其置于磁力搅拌恒温水浴锅中进行萃取。待萃取达到平衡后,将萃取纤维从血清瓶中取出并插入GC进样口,进行仪器分析。SPME优化过程所使用的标准溶液质量浓度为100.0 ng/L。
2 结果与讨论
2.1 萃取材料的优化
由于UiO-66/HOCN复合材料中HOCN掺杂含量的改变会直接影响该复合材料对目标分析物的萃取效率,因此先通过预实验来探究其对农药的吸附性能和机理。如图1所示,当UiO-66/HOCN复合材料中HOCN占PTA的质量分数为50%时(UiO-66/HOCN-50),对9种农药(OCPs和PYs)的萃取效果最优。随着HOCN掺杂含量的继续增加,复合材料的萃取效率逐渐降低。此实验现象可能是因为随着HOCN掺杂含量的增加,所得到的UiO-66/HOCN复合材料与目标物之间的π-π堆积相互作用和静电相互作用逐渐增强,使得对目标物的萃取效率逐渐增高。当HOCN掺杂含量继续增加时,可能由于堵塞了UiO-66的孔道,影响了UiO-66对目标物的萃取性能,从而降低了复合材料对目标物的萃取效率。因此,将HOCN掺杂质量分数为50%的UiO-66/HOCN复合材料用于之后的实验中。
此外,利用Zeta电位仪对UiO-66/HOCN与9种农药目标物(OCPs和PYs)之间的静电相互作用进行了表征,结果表明,UiO-66/HOCN的Zeta电位值为+8.73 mV,在吸附带负电的OCPs和PYs(混合溶液Zeta电位值为-6.32 mV)之后,电位值降至-2.18 mV,说明UiO-66/HOCN-50与9种农药目标物之间确实存在着静电相互作用。
2.2 UiO-66/HOCN复合材料的表征
首先,利用透射电子显微镜(transmission electron microscope, TEM)对UiO-66/HOCN复合材料的形貌进行了表征。如图2a所示,在薄层的HOCN纳米片上分布着形貌均一的UiO-66纳米颗粒,表明UiO-66/HOCN纳米复合材料的成功制备。此外,利用X射线衍射(X-ray diffraction, XRD)对UiO-66/HOCN的结构进行了表征。如图2b所示,所制备的UiO-66/HOCN复合材料同时具备UiO-66纳米颗粒和HOCN纳米片的共同特征峰(7.34°、8.56°和25.72°),再次表明成功地制备了UiO-66/HOCN复合材料。
同时,通过氮气吸附-脱附实验对UiO-66/HOCN-50的比表面积和孔隙率进行了表征(见图2c)。结果表明,UiO-66/HOCN-50复合材料的BET比表面积为725.8 m2/g,相比于HOCN的比表面积(BET=220.5 m2/g), UiO-66/HOCN复合材料的比表面积有了很大的提高。高比表面积有利于萃取效率的提高,使得该复合材料适合作为一种SPME的涂层材料。此外,孔径分布实验结果表明UiO-66/HOCN-50同时具有微孔和介孔结构(见图2d),可以用于萃取不同尺寸大小的目标物分子。
2.3 UiO-66/HOCN复合材料的热稳定性和化学稳定性
在GC-MS热解吸过程中,固相微萃取纤维的热稳定性非常重要。通过热重分析法(Thermogravimetric analysis, TGA)对UiO-66/HOCN复合材料的热稳定性进行了表征。如图3a所示,所制备的UiO-66/HOCN复合材料具有良好的热稳定性,当温度达到500 ℃时,复合材料的重量只有约10%的损失。与纯UiO-66纳米材料相比较,UiO-66/HOCN复合材料的热稳定性提高了约10%。因此,UiO-66/HOCN复合材料优良的热稳定性能够满足GC-MS的热解吸过程。
此外,UiO-66/HOCN复合材料的化学稳定性也同样重要。通过将UiO-66/HOCN复合材料纤维浸入有机溶剂(甲醇、二氯甲烷和丙酮)和酸碱溶剂(pH=5.0和pH=9.0)中24 h,以此方法来探究UiO-66/HOCN复合材料纤维的化学稳定性。如图3b所示,在不同的溶剂中浸泡24 h后,纤维涂层的厚度和形态未发生明显变化,实验结果表明该复合材料纤维具有良好的化学稳定性。
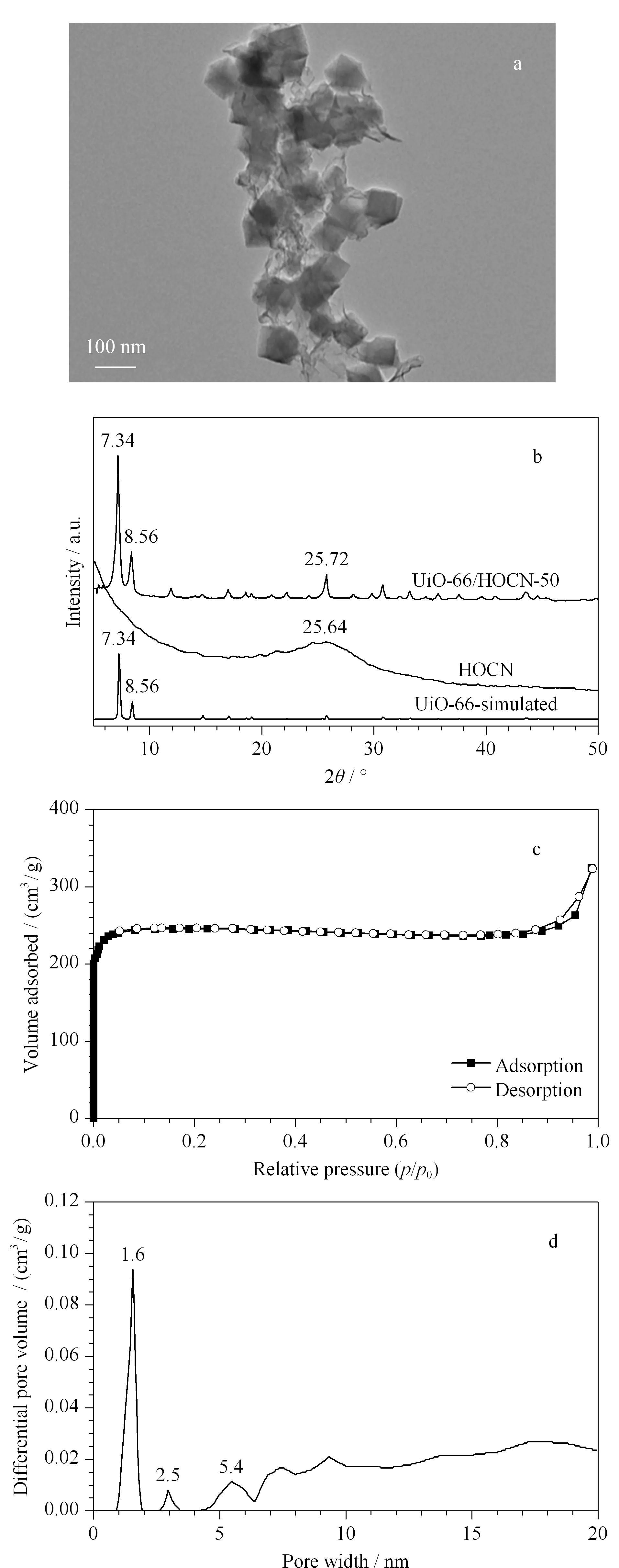
图 2 UiO-66/HOCN复合材料的(a)TEM、(b)XRD、 (c)等温氮气吸附-脱附以及(d)孔径分布表征图Fig. 2 (a) Transmission electron microscopy (TEM) image, (b) X-ray diffraction (XRD) pattern, (c) N2 adsorption-desorption isotherms, and (d) pore size distribution of UiO-66/HOCN composite materials
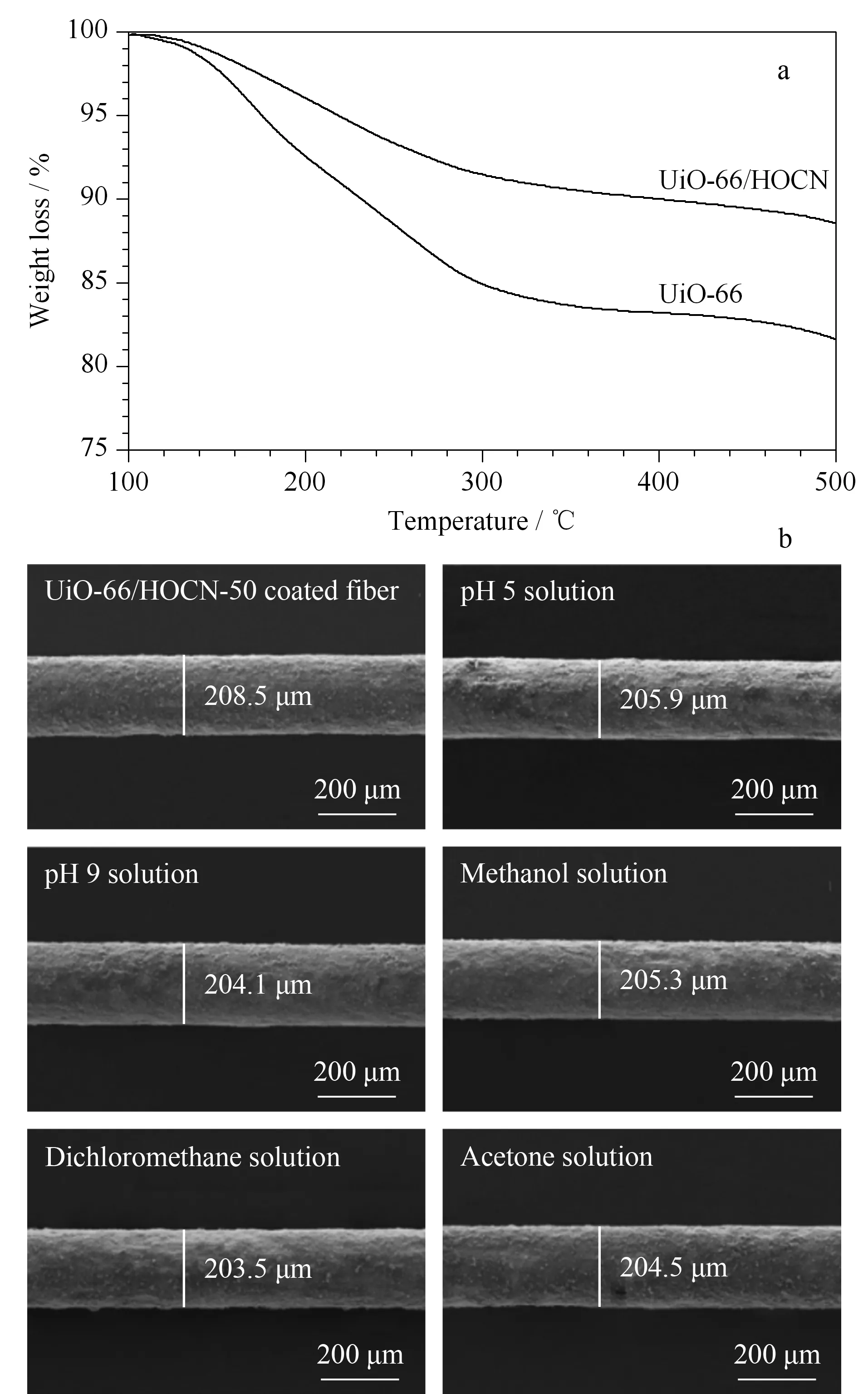
图 3 UiO-66/HOCN复合材料的(a)热稳定性表征图和 (b)化学稳定性表征图Fig. 3 (a) Thermal stability and (b) chemical stability of UiO-66/HOCN composite materials
2.4 SPME方法的优化
2.4.1萃取过程的优化
固相微萃取过程的主要影响因素有萃取温度、萃取时间和搅拌速率。首先,通过改变油浴锅的温度对萃取温度进行了优化。如图4a所示,随着萃取温度的升高,萃取效率也随之增高。当萃取温度达到50 ℃时,萃取效率达到最大值。然而继续增加萃取温度,萃取效率反而逐渐降低。这可能是由于随着萃取温度的提高,目标物分子的运动速率也随之增大,从而提高了萃取效率。然而,又因为吸附是一个放热过程,在萃取温度过高时,萃取效率反而降低。因此,最优的萃取温度为50 ℃,并用于之后的试验。
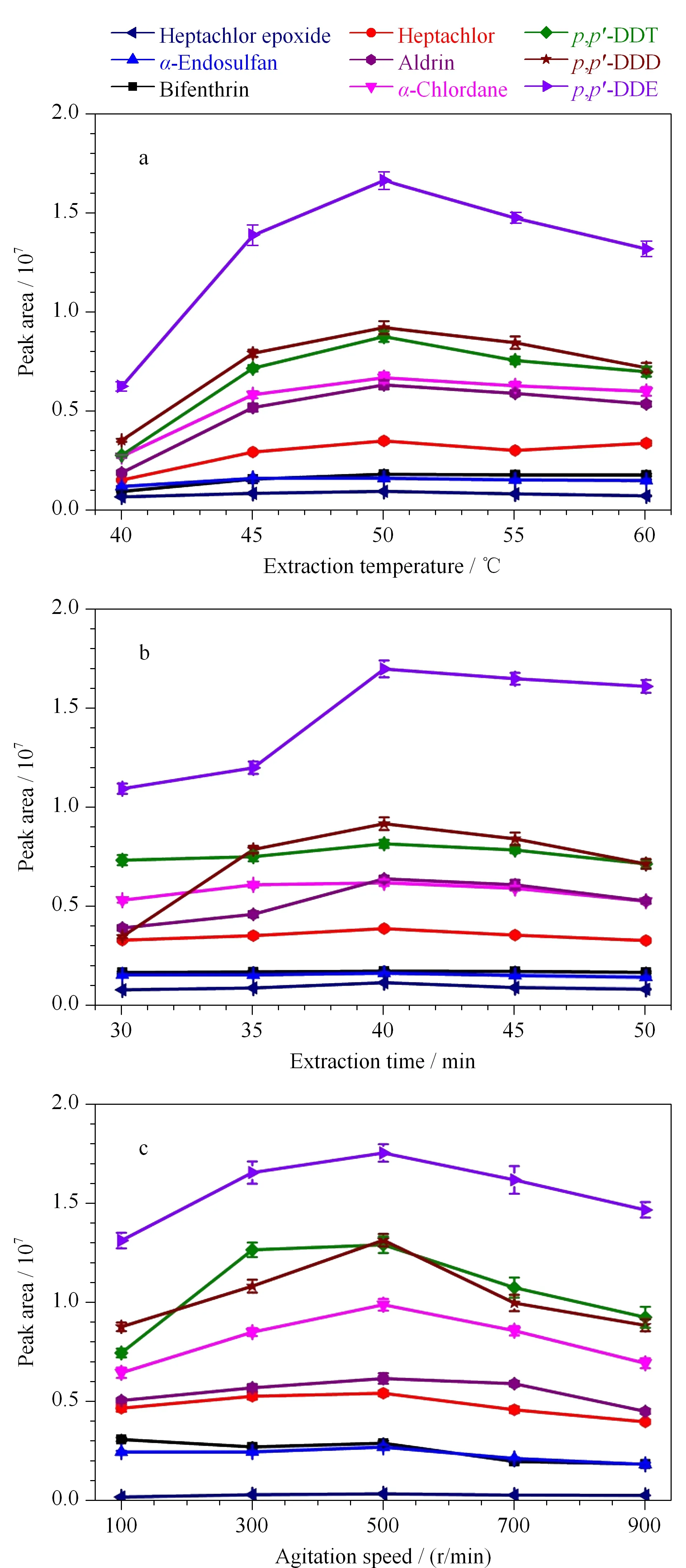
图 4 (a)萃取温度、(b)萃取时间和(c)搅拌速率对 9种农药萃取效率的影响(n=3)Fig. 4 Effects of (a) extraction temperature, (b) extraction time and (c) agitation speed on extraction efficiency of the nine pesticides (n=3) Experimental conditions were the same as those in Fig. 1. When one of the factors was investigated, all other factors remain the same.
此外,进一步考察了萃取时间为30~50 min时对萃取效率的影响。如图4b所示,随着萃取时间的延长,萃取效率也随之增加,当时间为40 min时,萃取达到平衡。因此,基于灵敏度和时间效率的综合考虑,将40 min设定为最优的萃取时间。同时,对搅拌速率也进行了优化。如图4c所示,随着搅拌速率的加快,萃取效率随之逐渐提高。然而,当搅拌速率超过500 r/min时,萃取效率逐渐减低。这是由于搅拌有利于加速目标物的扩散速率,促进目标物向涂层的迁移,从而提高萃取效率。但是,当搅拌速率太高时,所产生的涡流降低了目标物与吸附材料之间的有效接触。因此,最佳的搅拌速率为500 r/min,并用于之后的实验研究。

表 2 所建立方法的线性范围、相关系数、检出限以及精密度Table 2 Linear ranges, correlation coefficients (R), limits of detection (LODs), and precision of the proposed method
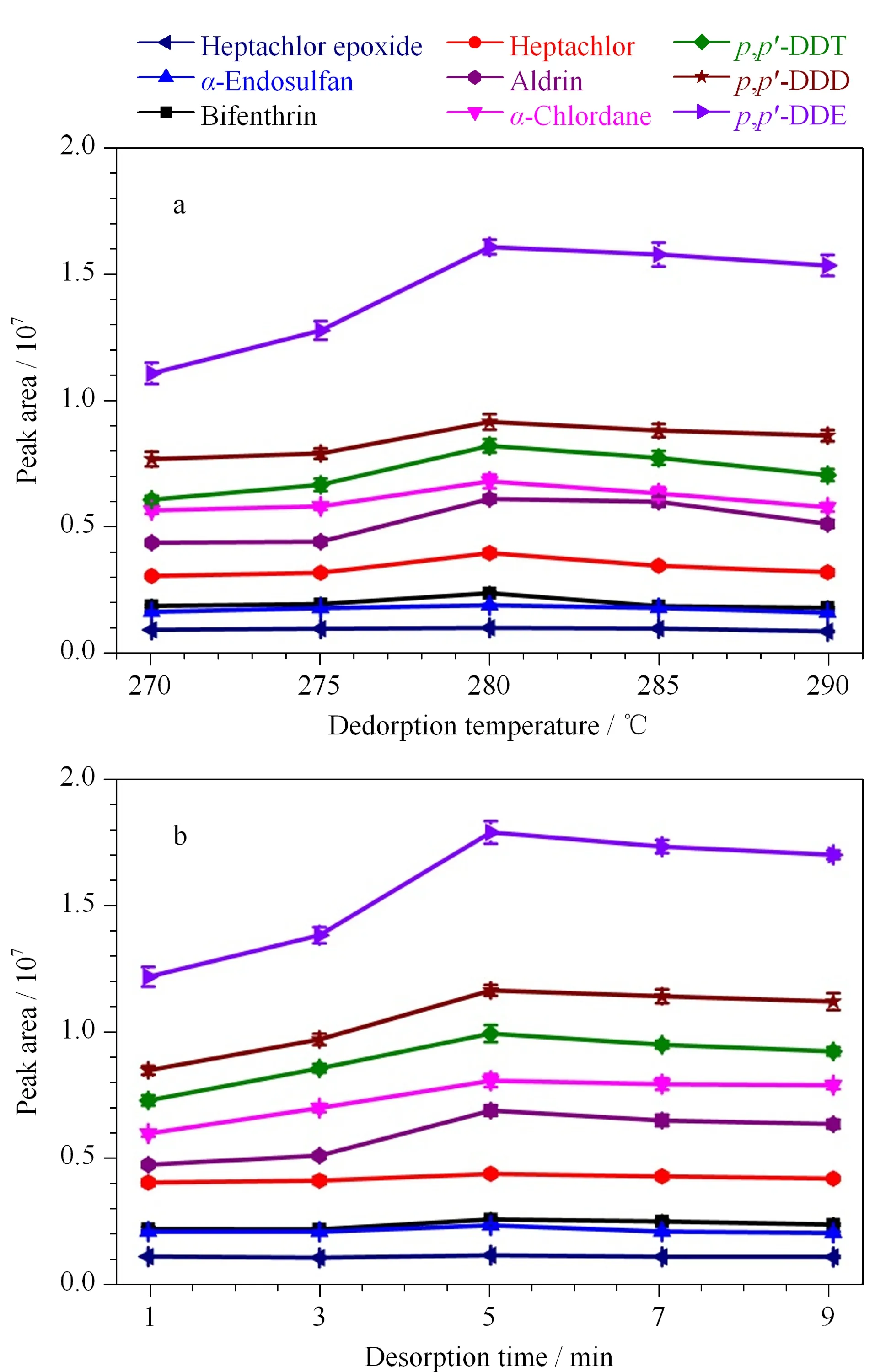
图 5 (a)解吸温度和(b)解吸时间对9种农药 萃取效率的影响(n=3)Fig. 5 Effects of (a) desorption temperature and (b) desorption time on extraction efficiency of the nine pesticides (n=3) Experimental conditions were the same as those in Fig. 1. When one of the factors was investigated, all other factors remain the same.
2.4.2热解吸过程的优化
为了获得良好的回收率,进一步研究了解吸温度在270~290 ℃范围内对萃取效率的影响,以确保目标物完全从纤维涂层中解吸。如图5a所示,随着解吸温度的升高,萃取效率逐渐增高。当解吸温度达到280 ℃时,萃取效率达到峰值。继续增高解吸温度,萃取效率没有明显变化。因此,最佳的解吸温度为280 ℃。此外,解吸时间也是影响萃取效率的重要因素之一。如图5b所示,当解吸时间为5 min时,萃取效率达到最大,继续增加解吸时间,则萃取效率基本保持不变。因此,最优的解吸时间为5 min,并用于之后的实验。
综上所述,最优的SPME条件如下:萃取温度50 ℃,萃取时间40 min,搅拌速率500 r/min,解吸温度280 ℃,解吸时间5 min。
2.5 检测体系的建立与验证
通过用丙酮稀释储备溶液来制备一系列浓度的标准溶液,用于标准曲线的绘制,每个浓度点重复3次。如表2所示,在最佳的SPME条件下,对9种农药目标物所建立的分析方法具有线性范围宽、相关系数良好、检出限(LODs)低的优点,能够满足痕量农药的分析。
此外,在最优实验条件下,利用100.0 ng/L的标准溶液对所建立的分析方法进行日内和日间精密度考察。通过3次平行实验,计算单根纤维和不同批次纤维间的相对标准偏差(RSD)。如表2所示,单根涂层的日内和日间RSD值分别在3.3%~7.5%和4.5%~8.9%范围内,不同批次纤维间的RSD值在5.2%~9.7%范围内。实验结果表明UiO-66/HOCN复合材料涂覆的固相微萃取纤维具有良好的重复性和重现性,为所建立分析方法的准确度和精密度提供了保障。
2.6 与其他方法的比较
将针对农药残留所建立的分析方法与其他文献报道的方法进行比较。如表3所示,本研究中所建立的分析方法具有灵敏度高、线性范围宽、重现性良好等优点。与其他方法相比,本研究所建立的方法具有最低的检出限,其得益于UiO-66/HOCN复合材料对目标物的高萃取效率。此外,该复合材料良好的热稳定性和化学稳定性为所建立分析方法的准确度和精密度提供了保障。实验结果表明通过原位溶剂热聚合法制备的固相微萃取纤维适用于复杂基质中农药的痕量分析。
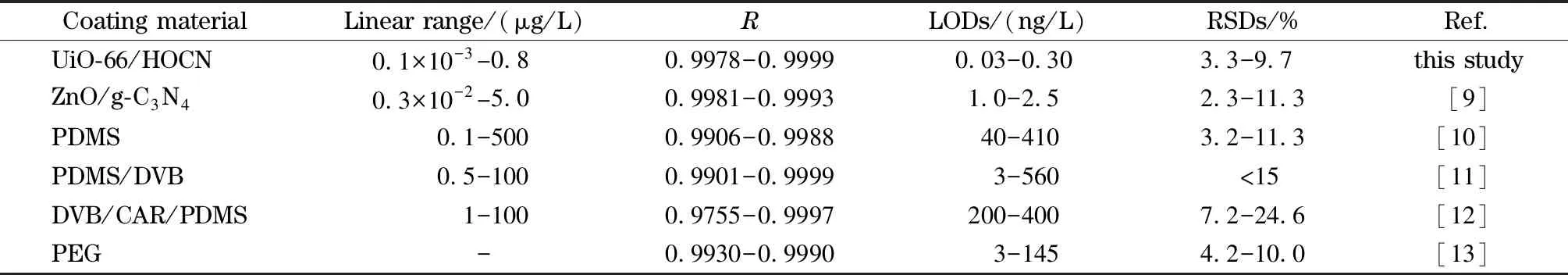
表 3 本文方法与其他已报道方法的比较Table 3 Comparison of the proposed method with other reported methods
PDMS: polydimethylsiloxane; DVB: divinylbenzene; CAR: carboxen; PEG: polyethylene glycol.

表 4 9种农药在实际红茶样品中的加标回收率及精密度(n=3)Table 4 Recoveries and RSDs of the nine pesticides spiked in a black tea sample (n=3)
* Not detected.
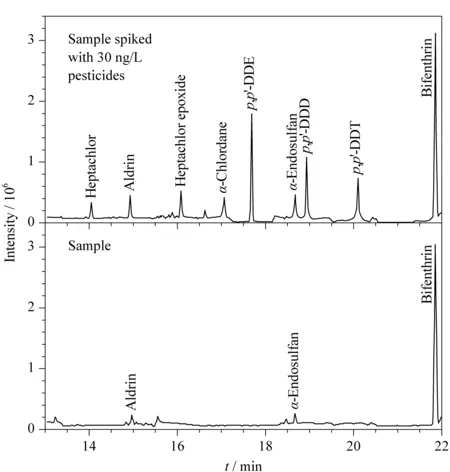
图 6 实际红茶样品和加标红茶样品的色谱图Fig. 6 Chromatograms of a black tea sample and a black tea sample spiked with standards Experimental conditions were the same as those in Fig. 1. Spiked level: 30 ng/L of each of the nine pesticides. Peak identifications: 1. heptachlor; 2. aldrin; 3. heptachlor epoxide; 4. α-chlordane; 5. p,p′-DDE; 6. α-endosulfan; 7. p,p′-DDD; 8. p,p′-DDT; 9. bifenthrin.
2.7 实际样品的测定
由于所建立的分析方法具有优良的性能,尝试将该分析方法用于实际红茶样品中9种农药的检测,并对该分析方法的回收率和精密度进行考察。如表4所示,所建立的分析方法成功地在实际红茶样品中检测出了艾氏剂(6.6 ng/L)、α-硫丹(54.7 ng/L)和联苯菊酯(185.8 ng/L)。此外,通过实际样品加标(8 ng/L、30 ng/L和100 ng/L)对所建立的分析方法的重现性和回收率进行了考察(n=3)。如表4所示,该分析方法的实际样加标回收率为82.9%~117.0%,并具有良好的重现性(RSD≤8.9%)。图6是实际样品和实际样品加标(30 ng/L)的色谱图。结果表明所建立的分析方法具有良好的准确度和精密度,适用于实际样品中农药残留的日常监测。
3 结论
在本研究中,将具有丰富活性位点的HOCN二维纳米材料与高比表面积的UiO-66相结合,利用原位溶剂热聚合法制备以UiO-66/HOCN复合材料为涂层的固相微萃取纤维,该纤维表现出良好的热稳定性和化学稳定性;将其与SPME-GC-MS相结合,建立了同时测定茶叶中9种农药残留的分析方法。UiO-66/HOCN复合材料的微-介孔结构及与目标物之间的π-π相互作用和静电相互作用在对目标物吸附的过程中发挥了重要作用,对9种农药表现出良好的萃取效率。在最优的SPME条件下,所建立的分析方法对9种农药目标物表现出了令人满意的灵敏度、回收率和重现性,具有检出限低、线性范围宽和线性相关系数良好等优点。将所建立的方法用于实际红茶样品中农药残留的检测,成功地在实际样品中检测出了艾试剂、α-硫丹和联苯菊酯。综上所述,所建立的分析方法适用于复杂基质中农药残留的分析和监测。
猜你喜欢
青少年科技博览(中学版)(2022年11期)2023-01-07
建材发展导向(2022年2期)2022-03-08
少儿科技(2022年2期)2022-03-05
北京航空航天大学学报(2021年9期)2021-11-02
建材发展导向(2021年14期)2021-08-23
重型机械(2020年2期)2020-07-24
民用飞机设计与研究(2020年1期)2020-05-21
中国航海(2019年2期)2019-07-24
纤维复合材料(2018年3期)2018-04-25
互联网天地(2016年2期)2016-05-04
