
经典名方甘姜苓术汤中甘草苷含量的HPLC 测定
2020-02-19 07:20:28贺丹彤邢春来成光宇
吉林中医药 2020年2期
刘 鹤,贺丹彤,邢春来,何 蕊,刘 睿,成光宇,位 鸿*
(1.长春中医药大学药学院,长春 130117;2 长春中医药大学附属医院中医药研究中心,长春 130021;3.长春中医药大学创新实践中心,长春 130117)
甘姜苓术汤出自《金匮要略·五脏风寒积聚病脉证并治第十一》[1],并被国家中医药管理局发布的《古代经典名方目录(第一批)》收录,全方由甘草、干姜、茯苓、白术组成,在治疗类风湿性关节炎、盆腔炎、肾积水等方面具有良好效果[2-5]。甘草苷是甘姜苓术汤中有效且稳定存在的成分,本实验建立了甘姜苓术汤中有效成分甘草苷的含量测定方法,并进行方法学验证[6-9],为甘姜苓术汤的质量控制提供实验依据。
1 方法
1.1 仪器与试药
1.1.1 仪器 美国Waters 公司2695 液相色谱仪,PDA 检测器;KH-250DB 型数控超声波清洗器(昆山禾创超声仪器有限公司);CP225D 型电子天平(德国sartorius 公司);Labconco Free Zone 冷冻干燥机(美国Labconco 公司)。
1.1.2 药品与试剂 甘草苷对照品(批号:111610-201607,供含量测定用,规格:20 mg,购于中国食品药品检定研究院);乙腈、乙醇为色谱纯,水为超纯水;其它试剂均为分析纯。
1.2 色谱条件与系统适应性 岛津WondaSil C18(4.6 mm×250 mm,5 μm)色谱柱;流动相乙腈(C)-0.05%磷酸水溶液,流动相比例0~8 min:19%C;8~45 min:19%~68%C;45~47 min:68%~90% C;47~55 min:90%C;55~60 min:90%~19%C;60~65 min:19% C;检测波长237 nm;流速1.0 mL/min。进样量10 μL;甘草苷的理论塔板数不低于5 000;柱温为室温。
1.3 对照品溶液的制备 精密称取甘草苷对照品,加70%乙醇制成每1 mL 含0.02 mg 溶液,摇匀,即得。
1.4 供试品溶液的制备 按处方量取甘草28.0 g,白术28.0 g,茯苓56.0 g,干姜56.0 g(共计168.0 g),加1 000 mL 水,浸泡30 min,明火煎煮至沸腾转为文火,煎至得煎液600 mL,冷冻干燥,得提取物粉末备用。
取提取物粉末(过四号筛)约0.1 g,精密称定,置10 mL 量瓶中,加70%乙醇8 mL,超声处理(功率250 W,频率40 kHz)40 min,放冷,用70%乙醇稀释至刻度,摇匀,0.45 μm 滤膜滤过,即得。
2 方法学验证
2.1 专属性考察 以十八烷基硅烷键合硅胶为填充剂(色谱柱型号:Inertsil ODS-C18(4.6 mm×250 mm,5 μm);以乙腈-0.05%磷酸水为流动相,梯度洗脱;检测波长为237 nm[10]。
分别精密吸取供试品溶液、对照品溶液、阴性对照溶液、溶剂各10 mL,注入液相色谱仪,甘草苷分离度大于1.5,理论塔板数大于5 000,色谱图见图1、2、3、4。
2.2 线性关系考察 精密吸取甘草苷对照品溶液2.0、4.0、8.0、10.0、15.0、20.0 mL,注入液相色谱仪,测定,以进样量(mg)为横坐标,峰面积积分值为纵坐标,绘制标准曲线[11]。回归方程:Y=1 307.6X-2 861.5,r=0.999 7(甘草苷),甘草苷线性范围为40.02~400.20 mg,试验结果见表1、图5。
图1 溶剂专属性考察
图2 甘草阴性专属性考察
图3 甘草苷对照品专属性考察

图4 供试品专属性考察

表1 线性关系考察
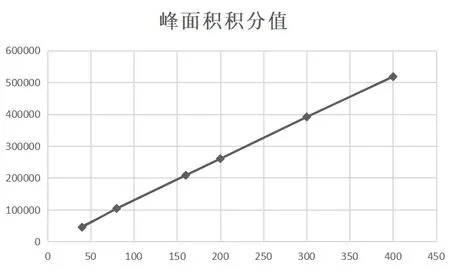
图5 甘草苷标准曲线
2.3 稳定性考察 精密吸取对照品溶液和同一批供试品溶液各10 mL,分别在0 h,2 h,4 h,8 h,12 h,24 h 进样,依法测定[12],以甘草苷峰面积积分值为指标,计算甘草苷RSD为0.43%,表明样品中稳定性良好,24 h 内测定结果可靠,结果见表2。
2.4 精密度考察 精密吸取甘草苷对照品溶液10 mL,注入液相色谱仪,连续进样6 次,依2.3 项下测定[13],记录峰面积积分值,计算得甘草苷对照品溶液连续测定的RSD 为0.74%,表明仪器、方法的精密度良好,结果见表3。
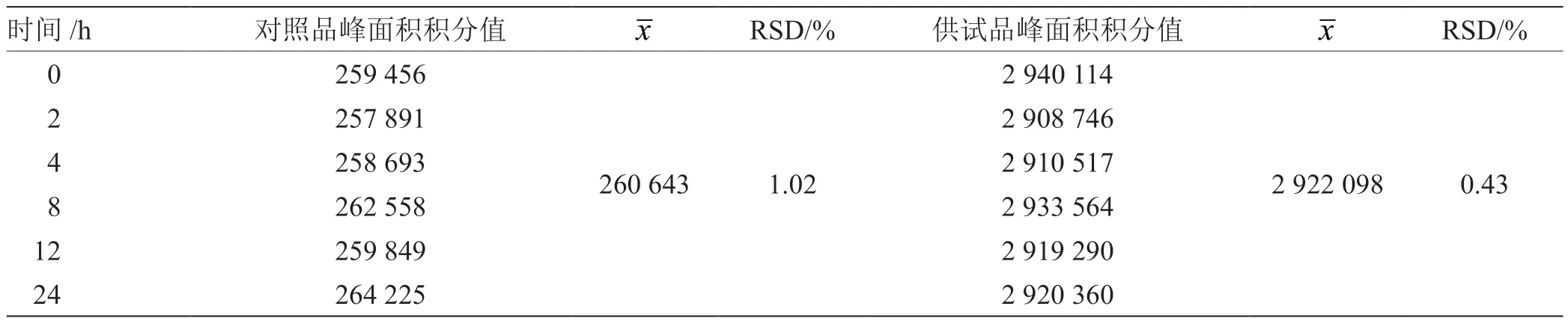
表2 甘草苷稳定性试验
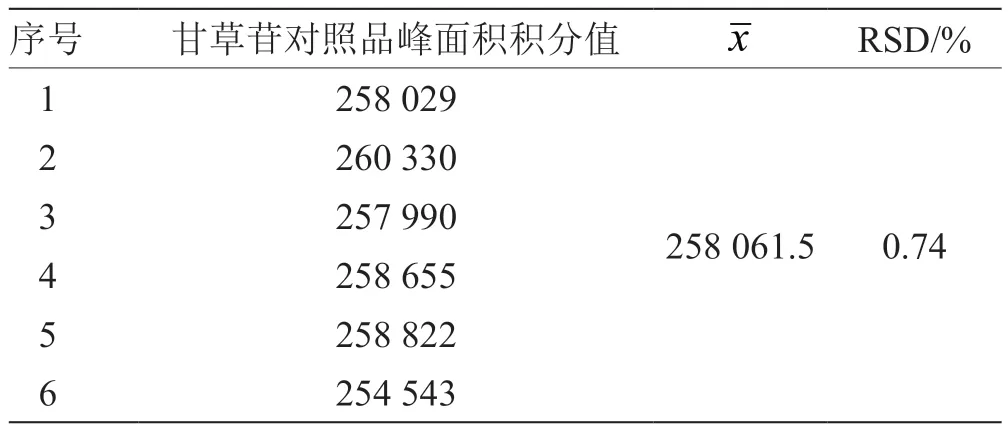
表3 甘草苷精密度试验
2.5 重现性考察 精密称取同一批样品共6 份,按2.2供试品溶液项下制备,依法独立测定[14],以甘草苷为含量测定指标,计算样品含量,结果RSD 为1.08%,平均含量为21.479 6 mg/g,表明该方法重现性良好,结果见表4。
2.6 准确度试验 取重复性试验同批样品,取约0.05 g,共6 份,精密称定,置10 mL 量瓶中,分别精密加入甘草苷对照品1.001 mg,加入70%乙醇,超声处理(功率250 W,频率40 kHz)40 min,放冷,用70%乙醇稀释至刻度,摇匀,0.45 μm 滤膜滤过,即得供试品溶液,依法测定[15],计算平均回收率为99.74%,RSD 为1.60%,表明本法准确性较好,方法可行,结果见表5。

表4 重现性试验
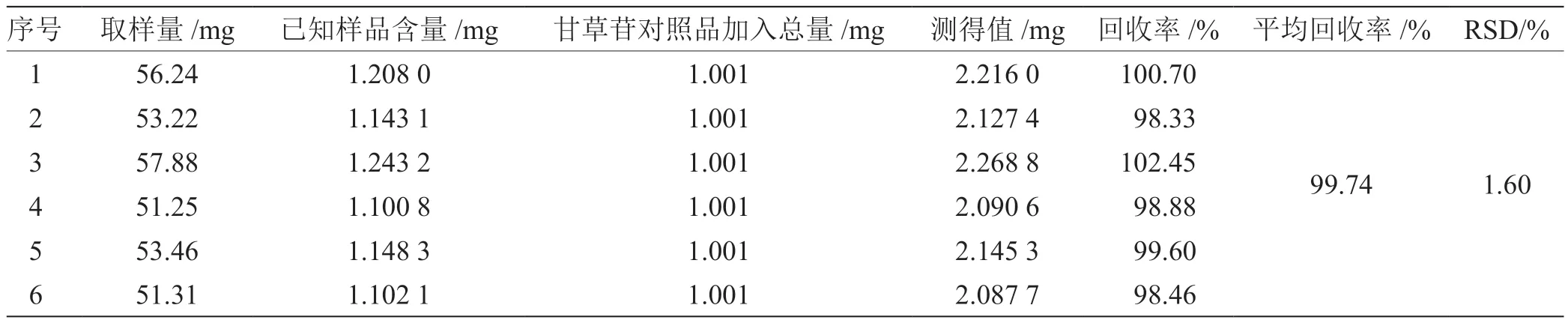
表5 回收率试验结果
2.7 含量测定 取不同产地不同批次药材饮片制备15批甘姜苓术汤提取物粉末,依法制备供试品溶液,分别精密吸取对照品溶液及供试品溶液各10 mL,注入液相色谱仪,测定峰面积,计算15 批甘姜苓术汤提取物粉末中甘草苷含量[16],结果见表6。
3 讨论
3.1 流动相的选择 本实验考察对比了不同的流动相系统[17],包括乙腈—水、乙腈-0.05% 磷酸水溶液;结果表明,以乙腈-0.05% 磷酸水溶液体系为流动相,采用梯度洗脱,能够达到较好的洗脱效果,各色谱峰的峰形和分离度相对较好,故选用乙腈-0.05% 磷酸水溶液作为流动相系统。
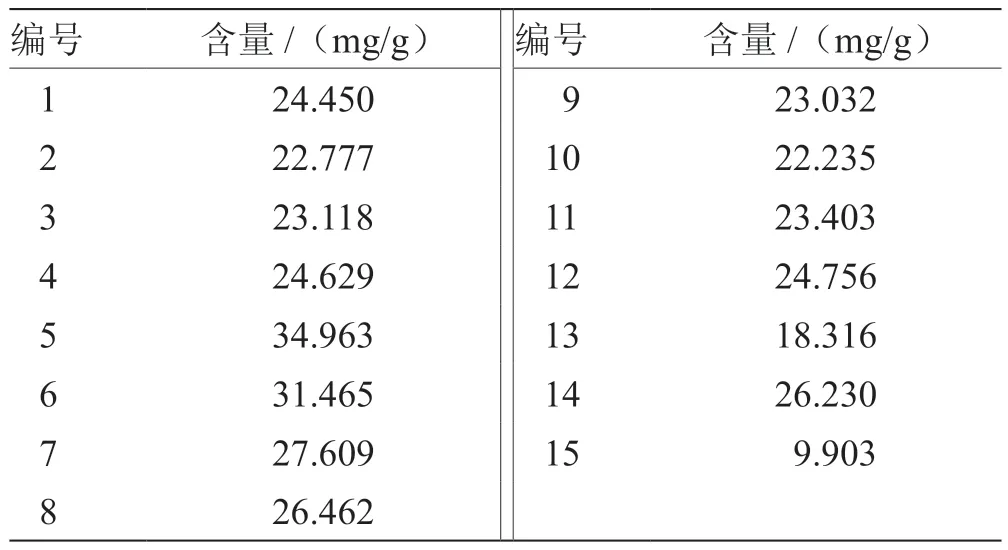
表6 测定结果
3.2 检测波长的选择 精密吸取甘草苷对照品溶液10 mL,进样分析,利用 PDA 检测器进行全波长扫描[18],测得最大吸收为237 nm,与2015版《中国药典》“甘草”项下收载的检测波长为237 nm 相符,因此选择237 nm为检测波长[19-20]。
3.3 结论 该方法建立了应用高效液相色谱法测定甘姜苓术汤中甘草苷含量的方法,该方法专属性好,精密度、稳定性、加样回收率均符合方法学验证要求,可以作为甘姜苓术汤质量控制方法。
猜你喜欢
基层中医药(2021年3期)2021-11-22 08:08:04
食品安全导刊(2021年21期)2021-08-30 08:21:40
中老年保健(2021年9期)2021-08-24 03:51:00
科学与财富(2021年35期)2021-05-10 11:54:37
食品安全导刊(2020年14期)2020-12-04 20:19:39
食品安全导刊·中旬刊(2020年5期)2020-06-04 08:43:53
艺海(2018年2期)2018-08-29 00:04:46
科学与财富(2017年32期)2017-12-20 22:13:54
科学与财富(2017年29期)2017-12-20 20:59:54
学术论坛(2016年5期)2016-05-17 05:44:43
