
强直性肌营养不良1例报告
2020-02-19 04:56高伟芳陈蓓蕾
中风与神经疾病杂志 2020年1期
高伟芳, 陈蓓蕾, 朱 艳
强直性肌营养不良(Myotonic dystrophy,DM)在临床上属于比较罕见的常染色体显性遗传的累及多系统的肌肉疾病。通过临床症状和体征,结合肌肉活检、基因检测可能有助于本病的鉴别诊断。强直性肌营养不良(DM)在临床上属于比较罕见的常染色体显性遗传的累及多系统的肌肉疾病。通过临床症状和体征,结合肌肉活检、基因检测可能有助于本病的鉴别诊断。
1 临床资料
患者女,44岁,因“双下肢乏力30 y,加重20 d”入院。患者30 y前感双下肢乏力,行走或爬楼梯后双下肢酸胀明显,休息后好转,双手用力握拳后不能立即伸开,未予重视。近20 d患者爬楼梯后自觉双下肢酸胀较前加重,为求进一步诊治,来我院风湿科就诊。追问家族史,患者诉其母亲有四肢无力症状多年,患者的儿子及患者妹妹有双手用力握拳后不能立即伸开症状,患者舅舅肺癌,两位表姐已去世,其中一位表姐的女儿有智力障碍。查体:面容瘦长,全身皮肤无红斑,无龋齿,无猖獗齿,无口腔溃疡,颞颌关节活动无受限。双手叩击时可诱发肌肉痉挛,出现肌球,双侧腓肠肌压痛明显,余四肢关节压痛肿胀均(-),双下肢无可凹性水肿,四肢肌力V-级,四肢肌张力正常,双下肢腱反射减弱,病理征(-)。辅助检查:肌酶谱:天门冬氨酸氨基转移酶59 u/L,乳酸脱氢酶327 u/L,肌酸激酶366 u/L;HLA-B27阳性(158),免疫球蛋IgG 6.02 g/L;抗核抗体弱阳性;凝血常规、D-二聚体、补体C3、补体C4、甲状腺功能、肿瘤标记物、抗中性粒细胞胞浆抗体、抗肌炎抗体、ANA抗体谱、甲状旁腺激素、病毒三项、风疹病毒IgM、弓形体IgM、巨细胞病毒IgM、单纯疱疹病毒IgM、抗O、类风湿因子未见明显异常。唇腺活检:(唇腺)涎腺组织,内见少量淋巴细胞、浆细胞散在浸润。胸部CT:两肺多发小结节影,双侧腋窝多发淋巴结。骶髂关节MRI:双侧骶髂关节炎表现。骶骨左侧异常信号。骶骨右后方软组织内斑片状水肿信号。双侧附件囊肿可能,右侧者为巧克力囊肿可能性大。双下肢MRI:(1)双侧腓肠肌内侧部萎缩,信号不均。(2)双侧胫骨前肌水肿。超声心动图:LVEF61%,三尖瓣轻度返流。腹部彩超:胆囊壁不光,胆囊结石。妇科彩超:双附囊肿。心电图:(1)窦性心律;(2)T波改变。肌电图提示双侧胫前肌,拇短展肌呈肌源性损伤表现,可见肌强直电位,余所检神经传导、F波及肌肉未见异常。提示肌强直性肌病。肌肉活检示:(右胫前肌肉)镜下示横纹肌组织,部分取胞浆变淡,肌丝消失,萎缩明显,间质内可见少量淋巴细胞浸润。基因检测:受检者强直性肌营养不良1型DMPK基因(CTG)n三核苷酸重复数110(异常重复数参考值>50次)(见图1),有诊断意义。受检者强直性肌营养不良2型CCTG基因重复数为16/18(见图2),无诊断意义。
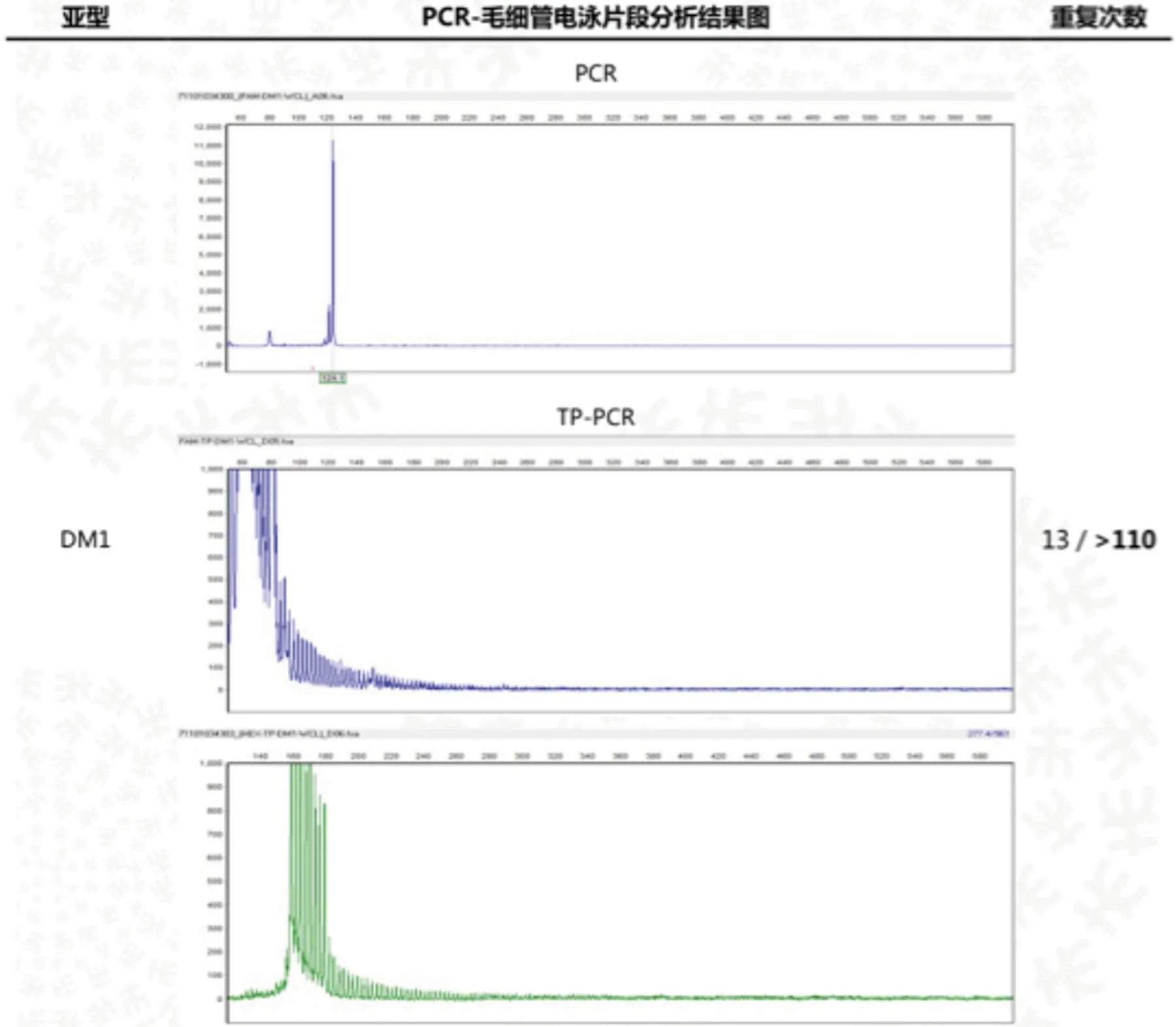
图1 DM1型基因检测
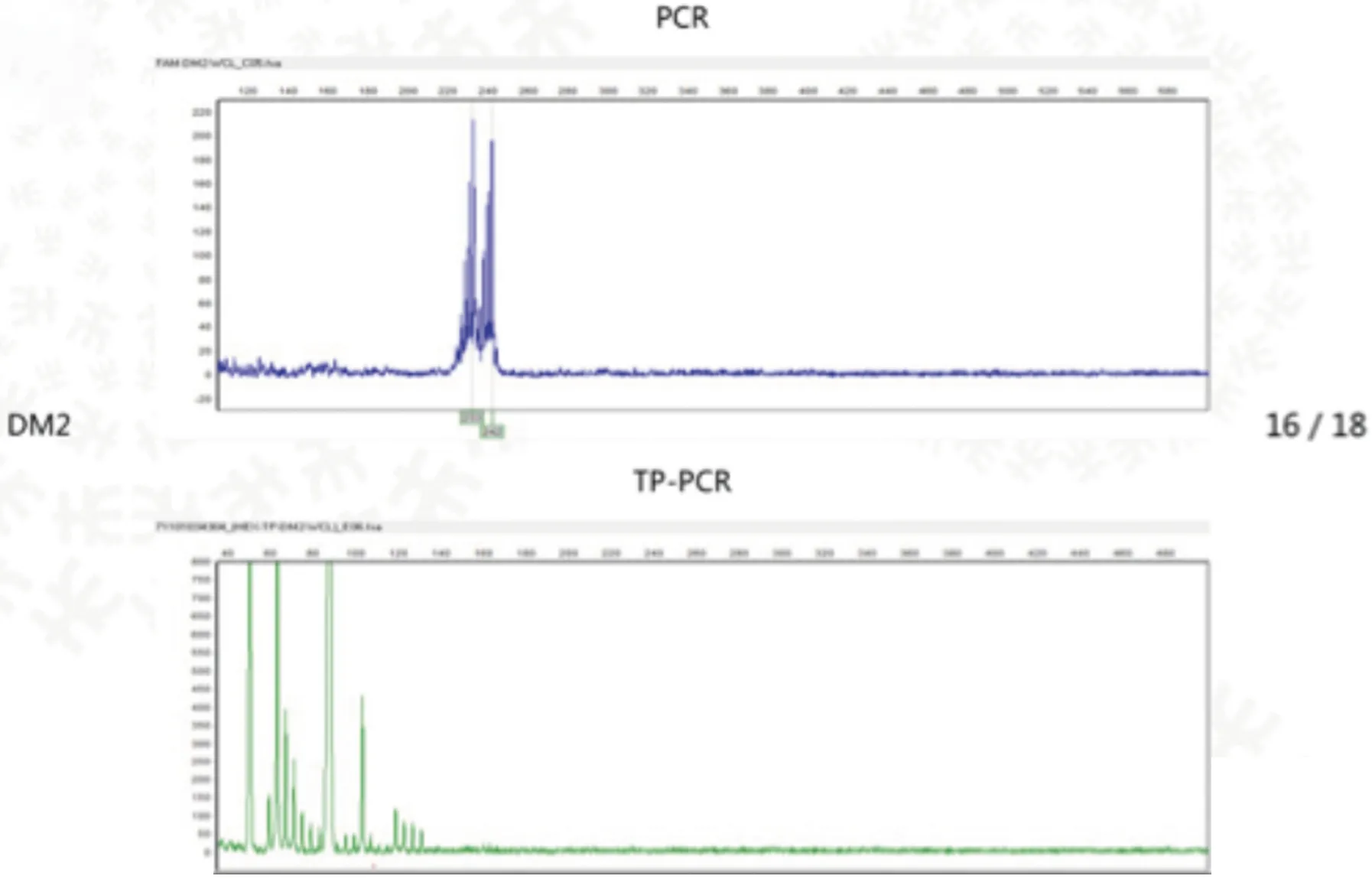
图2 DM2型基因检测
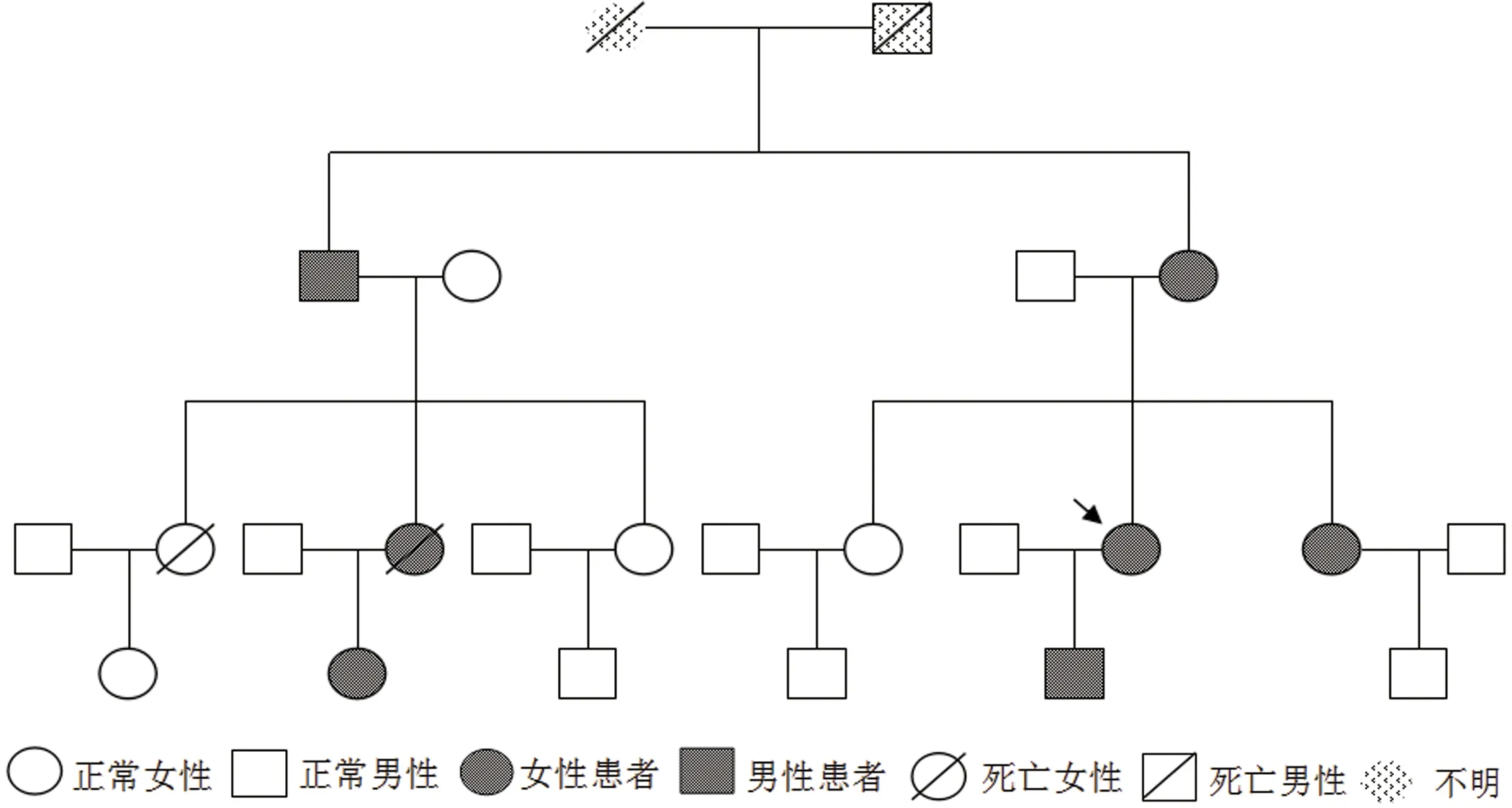
图3 家族系谱图
2 讨 论
强直性肌营养不良分为DMl和DM2两型,DM1型发病率高于DM2型,确诊依赖基因检测。DMl基因位点位于不稳定的CTG三核苷酸3’UTR的DMPK,基因定位于19q13.3,DM2基因位点位于不稳定的CCTG四核苷酸重复扩张的锌指9基因的内含子内,基因定位于3q2[1,2]。正常人CTG重复数在5~37之间,一般认为 CTG 重复扩增数目超过50即有意义[3]。依据基因检测,本例患者DMPK基因(CTG)n三核苷酸重复数110,DM1诊断明确。结合本患者就DM1型进行相关文献复习。
DM1临床表现多样,主要表现为肌强直、肌无力、肌萎缩,以及广泛的全身性病损,肌电图呈肌强直电位,肌活检主要特征是发现肌纤维大小不等,肌膜核增多、核内移,肌质块形成,Ⅰ型纤维萎缩和Ⅱ型纤维肥大,且肌质块为DM的一种特征性的肌肉病理改变[4]。肌电图在DM的临床诊断中非常重要,该患者初诊于风湿免疫科,肌电图显示肌强直电位,提示强直性肌病,遂进一步完善相关检查,肌肉活检提示肌肉萎缩。可见肌电图是初筛DM的一个重要手段。肌强直现象表现为肌肉起动困难,收缩后不能放松。肌强直首先多为肢体远端受累,如本患者及其子用力握拳后不能马上伸开,疾病进展过程中逐渐累及头面部及近端肌肉,可以出现吞咽困难、饮水呛咳、构音障碍,用力闭合眼睑不能立即睁眼,欲咀嚼时不能张口等。肌强直具有“热生现象”,即反复活动后强直症状减轻[5]。典型的肌强直易通过叩击鱼际肌或舌肌所引出,形成肌球征,被称为“叩击性肌强直”。患者双侧大鱼际肌球现象明显。肌无力及肌萎缩以肢体远端多见,其次为颈面部及四肢近端肌肉。颞肌及咬肌萎缩往往合并双眼睑下垂、脱发等构成强直性肌营养不良的独特的“斧头脸面容”,胸锁乳突肌萎缩时可呈“鹅颈”。DM患者可有轻度CK,LDH轻度升高[6]。患者辅助检查提示肌酶升高。骨骼肌以外的多系统受累是DM的重要临床表现。DM患者可出现各种心律失常甚至心肌病,主要以心脏传导异常发生率为高,约30%~75%,心脏受累为该病主要死因之一(约1/3)[7]。白内障也是本病常见临床症状,白内障极早出现的病例高达90%,但引起视力明显减退者较少。特征表现为后极部彩红样点片状浑浊,特征性白内障可作为强直性肌营养不良一个主要的甚至唯一的临床特征,为早期诊断提供重要临床依据[8]。呼吸系统受累主要是由于膈肌无力引起的咳痰无力及肺泡通气不足,再加上肌肉组织营养不良,患者横纹肌和平滑肌纤维丧失,常发生食管扩张、误吸,因而导致患者肺部感染反复发作[9]。强直性肌营养不良Ⅰ型患者的的早期死亡40%是由吸入性肺炎进而引起的呼吸衰竭造成。全身麻醉是造成呼吸衰竭的最常见诱因[10]。内分泌改变以性腺发育不良多见,男性患者可出现早秃、睾丸萎缩、性欲减退、阳痿,女性患者常见卵巢功能不全、月经失调、不孕和习惯性流产等,男性患者表现较女性患者严重。男性早秃是本病的突出体征之一[11]。糖代谢方面,多表现为胰岛素对糖负荷反应增高,出现糖耐量异常和高胰岛素血症,糖尿病的发病率不高[11]。中枢神经系统受累,磁共振上可见脑白质病变,两项意大利成人型DM1的系列研究显示66%~80.4%的患者表现为白质异常,其中颞前叶白质异常为39%~44%,此外国内MRI常有伴有颅骨增厚的报告,其可能也是DM1中枢性病变的一个影像学表现[12]。患者中枢神经系统受累可出现嗜睡、智能减低等临床表现,不同程度的日间嗜睡症状在强直性肌营养不良Ⅰ型患者中发生率39%以上[10],智力低下主要见于儿童型[13],患者表姐的女儿从小智力低下,可进一步基因诊断是否为强直性肌营养不良的儿童型。有文献报道DM1的遗传不稳定性可能增加患者肿瘤的发生率,常见并发肿瘤有上皮细胞瘤、胸腺瘤、甲状腺肿瘤、结肠癌等[14]。患者舅舅患有肺癌,考虑是不是与强直性肌营养不良有关。患者因双下肢乏力就诊,辅助检查提示肌酶升高,就诊于我院风湿免疫科,查骶髂关节MRI提示骶髂关节炎,查阅相关文献,未发现肌营养不良与骶髂关节炎的发生有相关联系,考虑是否肌强直、肌无力、肌萎缩多年,造成活动不便,磨损骶髂关节,从而导致骶髂关节炎,但并未发现有文献资料支持。
DM是常染色体显性遗传病,且有遗传早现现象,即代间传递时,(CTG)n拷贝数逐代增加,而发病年龄逐代提前,症状逐代加重[15]。临床上研究发现DM1型认知功能下降程度、心律失常严重程度与CTG重复扩增量呈正相关,与发病年龄呈负相关[13]。本例患者为先证者,虽然家族中其他人并未进行基因检测明确诊断,但依据患者所述,患者本家族发病规律符合常染色体显性遗传规律。
目前强直性肌营养不良尚无特殊疗法,且该病有遗传早现现象,应该引起我们高度重视,做到早期发现,及早诊治。
猜你喜欢
保健与生活(2022年13期)2022-07-06
疯狂英语·新阅版(2021年8期)2021-09-10
中华骨与关节外科杂志(2021年12期)2021-08-31
昆明医科大学学报(2021年6期)2021-07-31
中华养生保健(2020年5期)2020-11-16
云南医药(2020年5期)2020-10-27
北京广播电视报(2019年8期)2019-03-27
武警医学(2019年2期)2019-03-05
上海医药(2017年2期)2017-03-01
中国实用医药(2016年26期)2016-11-07
