
双醛纤维素碱性降解的研究*
2020-02-09 02:14王宝玉曾锦豪陈三龙
广州化工 2020年1期
王宝玉,曾锦豪,何 敏,陈三龙
(广东轻工职业技术学院,广东 广州 510300)
高碘酸钠氧化纤维素生成双醛纤维素,双醛纤维素低毒、可生物降解、生物相容性好,能够发生还原、氧化、磺化和胺化等衍生反应,特别是双醛纤维素衍生反应结合超声波或均质化处理能够分离出纳米微晶或纳米微纤纤维素,使其在生物医药、造纸、废水处理、化妆品等领域具有潜在的应用前景。然而双醛纤维素不稳定,在碱性室温条件下易产生β-烷氧基消除反应,生成半缩醛烷氧基阴离子,分子量和聚合度降低,最终生成乙醇酸和α,γ-二羟基丁酸[1]。学者们通过DAC的碱性水解研究高碘酸盐氧化纤维素的机理。Calvini等[2]通过测定碱性水解DAC的聚合度测定DAC中醛含量,避免了碱性水解对醛含量测定的影响,并支持高碘酸钠不均匀氧化的机理。在双醛纤维素复合材料的应用方面也应考虑DAC的碱性降解。如利用壳聚糖和DAC制备伤口敷料,在碱性条件下将Ag+氧化为Ag0[3]。高碘酸钠氧化纤维素生成纤维素珠粒,珠粒在碱性条件下不稳定,易破碎[4]。DAC交联壳聚糖吸附蛋白质,DAC碱性条件下不稳定[5]。
目前,有关双醛纤维素碱性水解的机理已经阐明,但对DAC碱性水解有关得率、结晶度的变化、形态和热稳定性的变化未见报道。本文主要研究中度氧化纤维素的碱性降解,利用FTIR、XRD、TGA、和SEM对DAC碱水解过程中化学基团、结晶度、形貌和热稳定性进行分析。
1 实 验
1.1 仪器与试剂
微晶纤维素购自上海阿拉丁生化科技股份有限公司(粒径50 μm)。高碘酸钠;氯化钠;乙二醇;氢氧化钠(分析纯)。
离心机,北京白洋医疗器械有限公司;tensor 27傅立叶变换红外光谱仪,Bruker;D8 Advance X射线衍射仪,Bruker;TG 209F1 Libra热重分析仪,Netzsch;EVO18扫描电镜,Carl Zeiss。
1.2 高碘酸氧化
参照Sirvio等的方法氧化微晶纤维素[6]。将4 g纤维素称重到250 mL锥形瓶中,加入200 mL蒸馏水和5.28 g NaIO4(摩尔比NaIO4/AGU=1:1 )和3.364 g氯化钠(摩尔比NaCl/AGU=7:3),并用铝箔反应容器覆盖,以防止光催化分解高碘酸盐。在水浴中于50 ℃搅拌反应3 h,反应结束后,加入乙二醇3 mL,继续反应10 min,离心产物洗涤数次至导电率接近纯净水的电导率(约40 cs/cm),用乙醇置换2次后真空干燥。
1.3 碱性水解
由于双醛纤维素在室温下能够发生快速降解[1],仅考虑水解时间和碱用量对水解的影响。共进行4组实验,碱性水解时间分别为DA15 min、DA220 min、DA340 min和DA460 min。碱用量为基于DAC醛基含量的用量,DA120%、DA250%、DA380%和DA4100%。测定每组降解样品的得率和醛基含量。
1.4 醛基含量的测定
根据ZHAO等[7]的方法测定醛含量。用乙酸将25 mL的0.25 M盐酸羟胺的pH值调节至3.2,加入100 mg(绝干质量)的未干燥的双醛纤维素,在室温搅拌2 h,过滤。用0.01 M氢氧化钠溶液滴定滤液,使pH值为3.2,同时测定双醛纤维素的质量。醛基的含量(mmol·g-1)计算如下:
醛基含量=[(V-V0)×C]/m
式中:V——滴定双醛纤维素所用氢氧化钠溶液体积,mL
V0——滴定纤维素所用氢氧化钠体积,mL
C——氢氧化钠摩尔浓度,0.01 M·L-1
m——双醛纤维素质量,g
1.5 样品表征
红外光谱采用KBr压片法制样,在4000~400 cm-1范围扫描32次,分辨率为4 cm-1。X射线衍射源为Cu-Kα(波长1.5406 nm),扫描步长0.0130°,衍射角2θ范围10°~40°。利用峰高法计算样品结晶度[8],见式(1)。热重分析在氮气氛围下,测试的温度范围30~700 ℃,升温速率10 ℃·min-1。采用扫描电镜分析样品的形貌,扫描前喷金。

(1)
式中:I002——002晶面(2θ=22.6°)的衍射强度
Iam——无定形区(2θ=18°)的衍射强度
2 结果与讨论
2.1 高碘酸钠氧化和碱性水解
通过高碘酸盐氧化在C2和C3位引入醛基,生成双醛纤维素。研究表明金属盐有助于缩短反应时间,提高氧化效率[6]。因此氧化反应加入氯化钠。氧化反应的得率为67.3%,醛含量为4.425 mmol·g-1。氧化后得乳白色悬浮液。离心后,经乙醇洗涤和真空干燥后,获得蓬松样品。
DAC随着碱性降解程度的增加,溶液颜色从DA1的淡黄色到DA4的棕色。随着降解时间和碱用量的增加,得率和醛含量降低,如图1所示。DA1虽然仅降解5 min,碱用量为20%,然而其得率为46.8%,醛含量为0.22 mmol·g-1。表明碱性降解是快速反应,也间接表明了醛基在纤维素分子链中的不均匀分布。
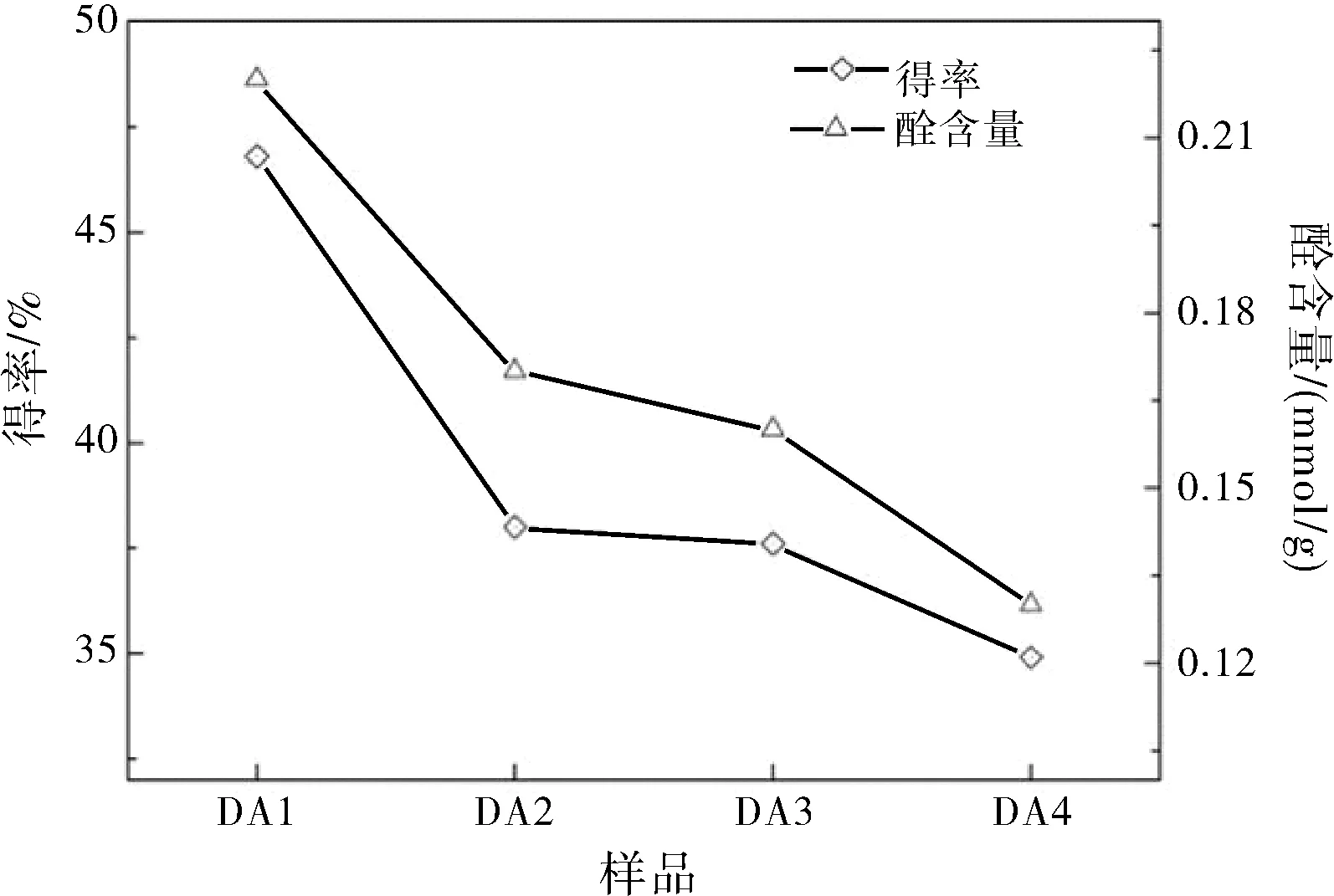
图1 DAC碱性降解的得率与醛含量
2.2 红外光谱分析
MCC、DAC及DA1~DA5的红外光谱如图2所示。3416 cm-1的宽峰归因于-OH伸缩振动[9],2903 cm-1处的强吸收归属于C-H的对称振动,1640 cm-1处的峰来源于吸附水。1430、1372和1317 cm-1处的峰分别为-CH2对称弯曲振动、C-H弯曲振动和-OH弯曲振动[10],1163、1033和896 cm-1处的峰分别归属于C-H伸缩振动,CH2-O-CH2伸缩振动和β-糖苷键振动[11]。在1059 cm-1处的峰为吡喃葡萄糖单元的C-O-C伸缩振动[12]。纤维素氧化后在出现了双醛纤维素的特征峰位,分别是归属于醛基的峰位1730 cm-1和归属于半缩醛的峰位891 cm-1[13],而且1059、1103和1317 cm-1处的峰强度弱于MCC。表明高碘酸钠将纤维素的羟基氧化为醛基。当DAC经碱性降解后,DA1、DA2、DA3和DA4在1730 cm-1的峰消失,半缩醛的峰位891 cm-1的峰移至896 cm-1。表明基于β-烷氧基消除反应的碱性降解使DAC的醛基消失,无定形区溶出,未氧化的结晶区得以保留,导致DA1,DA2,DA3和DA4的光谱与MCC相同。
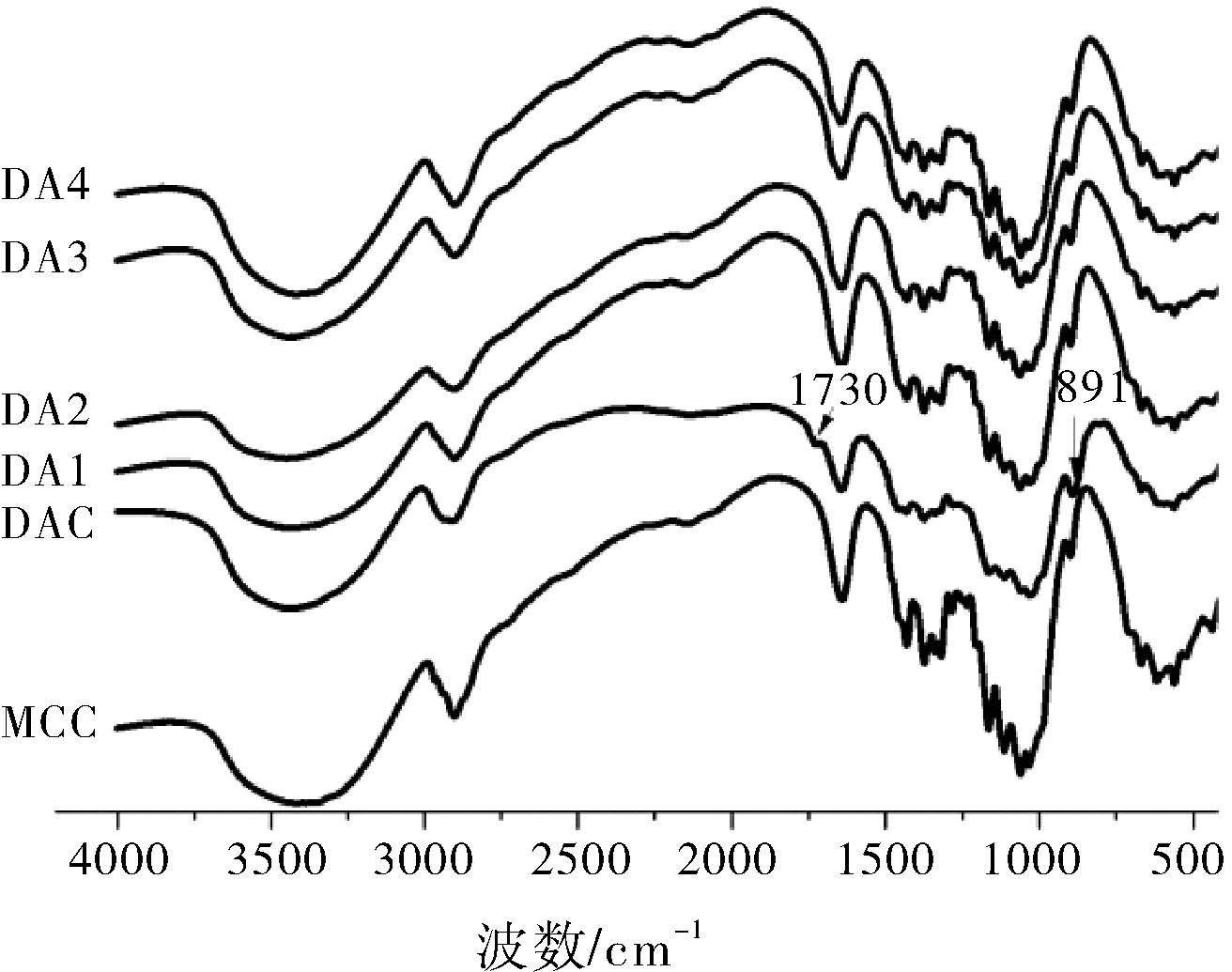
图2 样品的红外光谱
2.2.1 表面形貌分析

图3 样品的SEM图像(放大倍数4000倍)
使用扫描电镜分析样品的形貌,如图3所示。微晶纤维素胞腔有裂隙,纤维表面的细纤维随机分布。胞腔塌陷。有纤维形态的,纤维态长度30~70 μm,宽8~20 μm。也有颗粒态的,颗粒态直径约20~30 μm。双醛纤维素的颗粒态增多,颗粒直径10~30 μm,纤维态长10~60 μm,宽6~13 μm。颗粒表面的微细纤维形态消失,颗粒呈收缩而非舒展状,较为紧密。表明氧化过程中由于半缩醛的生成使纤维素结构紧密。
碱水解使纤维素链断裂,部分纤维素溶出,DAC原结构被破坏,DA1、DA2和DA3呈聚集态颗粒,颗粒内部多孔。也存在另一种可能性,即碱性水解产生细小颗粒,细小颗粒再絮聚成疏松的颗粒。使用超声分散DA4,超声10 min,有部分颗粒尺寸小于100 nm颗粒。超声后悬浮液有清晰的光柱,丁达尔效应显著。超后静置24 h,产生絮聚,形成较大颗粒,颗粒稳定悬浮,呈云彩状。表明DAC的碱水解悬浮液不稳定,易絮聚。
2.2.2 结晶度
样品的X射线衍射如图4所示,由式(1)计算MCC、DAC、DA1~DA4的结晶指数分别为81.8%、41.0%、57.9%、58.7%、65.1%和68.3%。高碘酸钠氧化微晶纤维素,氧化先发生在无定形区,接着结晶区的界面,随着反应的进行,结晶区被逐渐氧化,导致结晶度从MCC的81.8%降至DAC的41%。碱性降解发生β-烷氧基消除反应,无定形区溶出,使结晶度随着碱性降解程度的增加而增加升高,即碱性降解在一定程度上恢复了结晶度。

图4 样品的X射线衍射图谱
2.2.3 热稳定性分析
利用热重分析研究样品的热稳定性能。样品的热重曲线和微商热重曲线分别如图5和图6所示。由图可知,所有样品的热降解分为二个阶段。
热降解过程中,由于结合水的蒸发,所有样品在48~62 ℃出现第一阶段的质量损失。其中,MCC的质量损失为4.72%,DAC质量损失为11.72%。碱性降解样品的质量损失介于MCC和DAC之间。样品结晶度的差异是导致质量损失主要原因。
所有样品在292~347 ℃间发生第二阶段快速质量损失,质量损失归因于纤维素分子链共价键断裂,纤维素分解为二氧化碳和水。其中微晶纤维素的最大降解速率下的温度为345.2 ℃,DAC为321.1 ℃。DAC结晶度低,易热降解,导致其最大热降解速率向低温方向移动。由于碱性降解使样品的结晶度增加,因此,碱性降解样品的最大热分解速率温度随着碱性降解程度的增加向高温方向移动,DA4的最大热分解速率的温度接近MCC。但DA1最大热分解速率下的温度为292 ℃,低于DAC的211.1 ℃。这是因为虽然DA1的结晶度高,但碱性降解使分子链断裂,分子链长度降低,分子链的数量增加。其最大热分解速率的温度较低是结晶度增加和分子链数量增加的共同作用结果。
DAC的热降解炭化残余质量为13.47%,大于微晶纤维素的炭化残余质量4.12%。所有碱性降解样品的炭化残余大于DAC。总之,DAC随着碱性降解程度的增加,其热稳定性增加。

图5 样品的TGA曲线
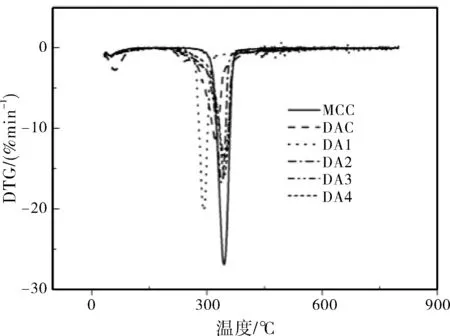
图6 样品的微商热重曲线
3 结 论
DAC的醛基在纤维素分子链上的不均匀分布,双醛纤维素碱性水解是快速反应,当用碱量为DAC醛基含量的20%水解5 min,得率为46.8%,醛含量为0.22 mmol·g-1。随着碱性水解程度的增加,得率和醛含量降低。碱性水解使纤维呈聚集态颗粒,颗粒内部多孔。随着无定形区的溶出,纤维素的结晶度增加,热稳定性改善。由于碱性水解得率低,纤维易絮聚,因此通过碱性水解分离纳米纤维素存在困难。可通过引入功能基团如羧基和磺酸基等,通过静电作用防止纤维颗粒的聚集。
猜你喜欢
陶瓷学报(2021年4期)2021-10-14
建材发展导向(2021年11期)2021-07-28
陶瓷学报(2021年1期)2021-04-13
中国有色金属学报(2018年2期)2018-03-26
制造技术与机床(2017年4期)2017-06-22
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
浙江农业科学(2016年11期)2016-05-04
文物保护与考古科学(2016年1期)2016-04-16
江苏农业科学(2015年1期)2015-04-17
科技创新导报(2014年34期)2015-01-13
