
反相高效液相色谱法测定盐酸左旋米那普仑原料药中多种杂质
2020-01-13 10:00周灵利陈维维张启余
成都大学学报(自然科学版) 2019年4期
周灵利, 陈维维, 张启余, 柯 潇
(成都康弘药业集团股份有限公司, 四川 成都 610037)
0 引 言
盐酸左旋米那普仑缓释胶囊作为成人重性抑郁障碍治疗的药品,于2013年经美国食品药品监督管理局(FDA)批准上市[1].盐酸左旋米那普仑作为该制品的原料药,通过选择性抑制5—羟色胺(5-HT)转运体和去甲肾上腺素(NE)转运体,从而抑制5-HT和NE的再摄取,且其对NE的再摄取抑制能力分别是度罗西汀和文拉法辛的27倍和17倍,具有较好的疗效.原料药的有关物质将直接影响药品的疗效和安全性,为了更好保障盐酸左旋米那普仑缓释胶囊的质量,有必要对其原料药的有关杂质进行控制.目前,国内仅有两篇该原料药异构体控制的文献报告[2-3],国外有关该原料药杂质控制的文献相对较多[4-6].但相关研究都不能完全涵盖该原料药中所需要控制的杂质.对此,本研究根据盐酸左旋米那普仑原料药的合成工艺和强制降解情况,确定需要控制的杂质(见图1),并采用反相高效液相色谱法测定盐酸左旋米那普仑原料药中的多种杂质,以对该原料药的质量进行控制.
1 材料与仪器
1.1 材 料
实验所用材料包括:盐酸左旋米那普仑,由成都康弘药业集团股份有限公司提供;
盐酸左旋米那普仑对照品(批号,PCL-#-M876)、杂质E(批号,PCL-#-MH14),购自成都迈斯克医药科技有限公司;杂质A(批号,2113-074AJ),购自TLC公司;杂质B(批号,L4X-103302-1411),购自CATO公司;杂质C(批号,20-MAR-14-01),购自QCC公司;杂质D(批号,16-02-1823),购自SINCO公司;三氟乙酸、甲醇、乙腈均为色谱纯试剂.
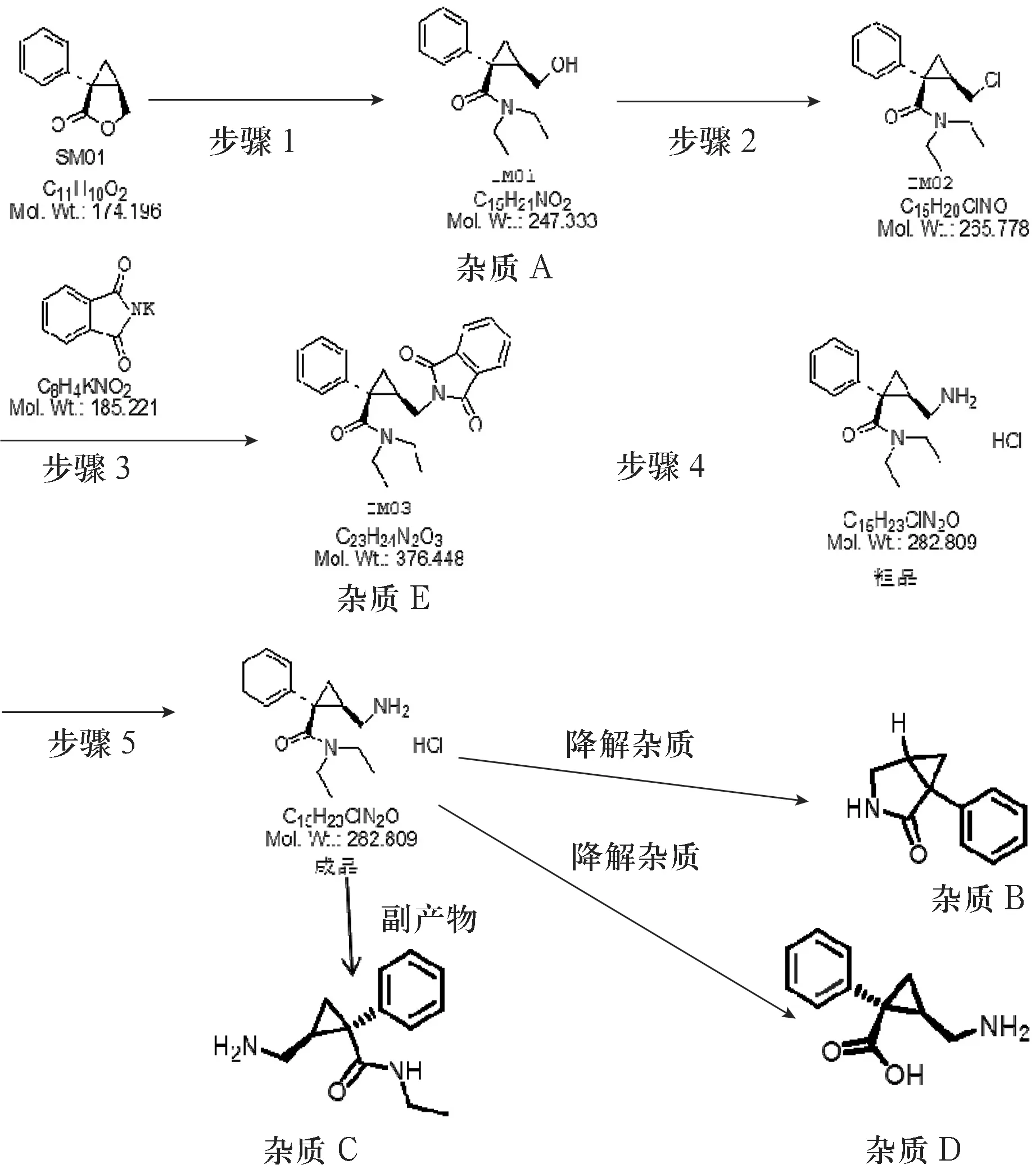
图1盐酸左旋米那普仑原料药杂质谱
1.2 仪 器
实验所用仪器包括:Ultimate-3000型高效液相色谱仪(赛默飞世尔科技中国有限公司),BP-211D型电子天平(赛多利斯科学仪器有限公司).
2 方法和结果
2.1 测定方法
2.1.1 色谱条件
实验的色谱条件为:色谱柱为Waters sunfire C18,5 μm,250 mm×4.6 mm;流动相以0.05%TFA水溶液为流动相A,以0.05%TFA乙腈溶液为流动相B,按表1程序进行梯度洗脱;柱温为30 ℃;流速为1.0 mL/min;检测波长220 nm;进样量10 μL.
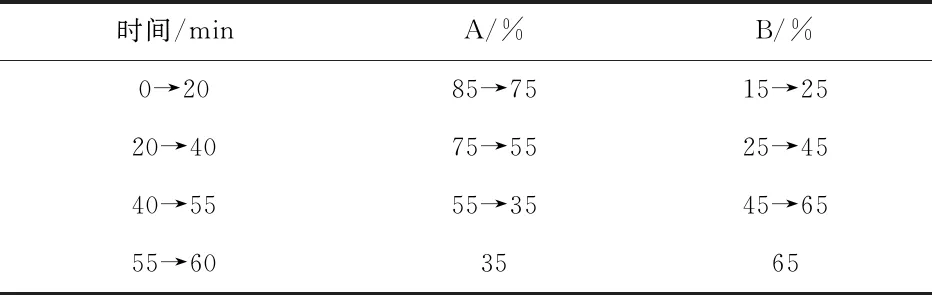
表1 梯度洗脱程序
2.1.2 溶液配制
1)供试品溶液.取供试品适量,加10%乙腈溶解并稀释制成每1 mL约含5 mg的溶液,摇匀,即得.
2)自身对照溶液.精密量取供试品溶液适量,加10%乙腈定量稀释制成每1 mL约含5 μg的溶液,摇匀,即得.
2.2 结 果
2.2.1 专属性
取盐酸左旋米那普仑有关物质A~E与盐酸左旋米那普仑标准品适量,配制每1 mL含有关物质A~E与盐酸左旋米那普仑各约含5 μg的混合溶液,作为系统适用性溶液;取盐酸左旋米那普仑有关物质A~E与盐酸左旋米那普仑标准品适量至不同的量瓶中,分别配制每1 mL约含5 μg的溶液,作为定位溶液;取以上溶液与10%乙腈(稀释剂)一起,按“2.1.1”项下色谱条件测定,记录色谱图,结果如图2所示.
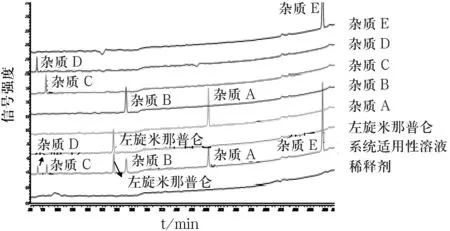
图2 专属性相关液相色谱图
由图2可知,各杂质之间以及杂质与盐酸左旋米那普仑之间分离度均符合要求.
2.2.2 破坏性
取盐酸左旋米那普仑原料药适量,加10%乙腈稀释成每1 mL约含盐酸左旋米那普仑50.0 mg/mL的溶液,作为储备液.取该储备液1 mL分别至8个10 mL量瓶中,按表2中的破坏条件分别加入适量处理溶剂,水浴80 ℃/光下处理相应时间,取出,放冷,加相应试剂中和后,10%乙腈(稀释剂)稀释至刻度,摇匀,作为破坏性实验相关溶液.取以上溶液按“2.1.1”项下色谱条件测定,记录色谱图,结果如图3所示.

表2 破坏条件
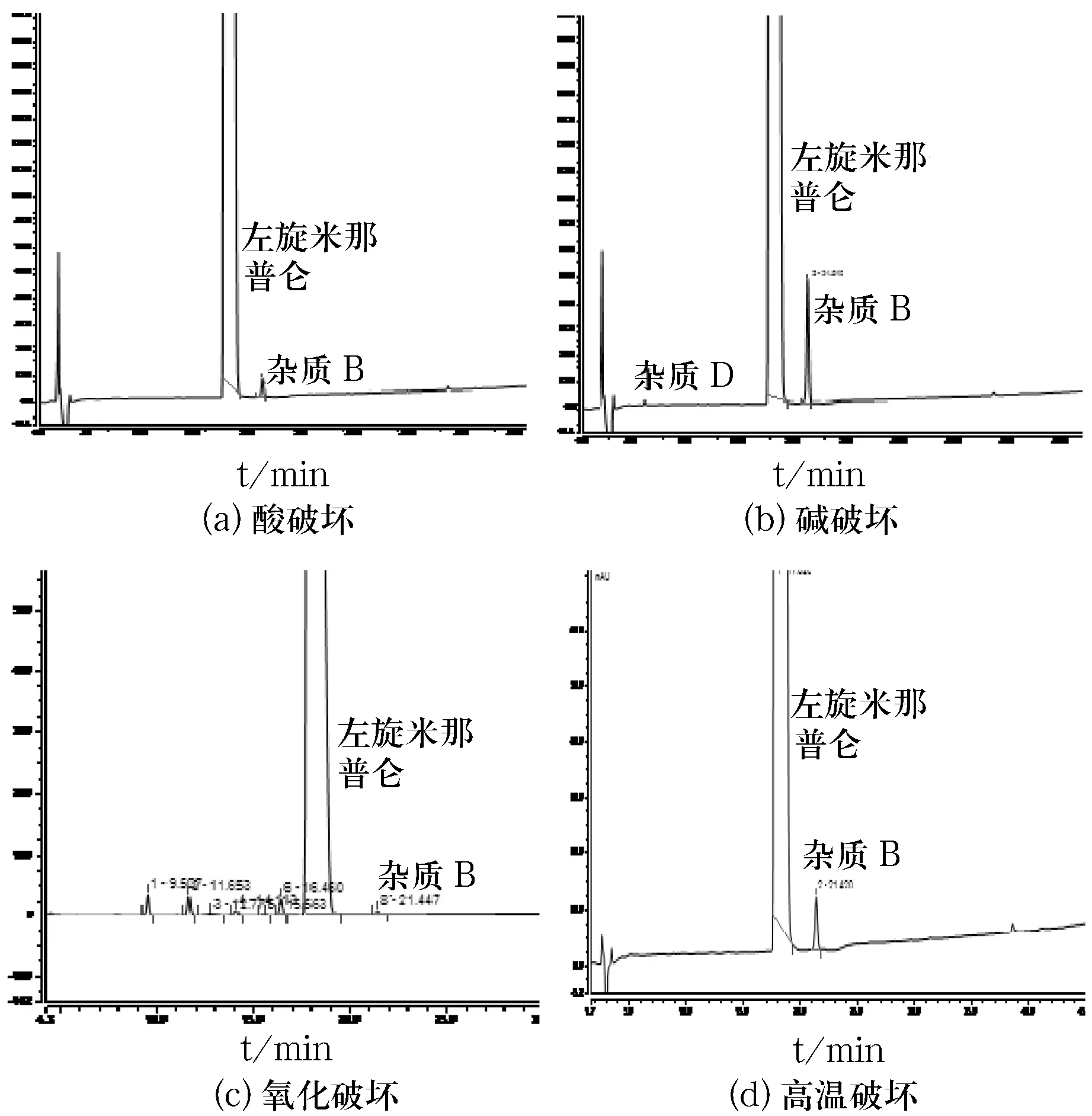
图3破坏性实验液相色谱图
由图3可知,样品在酸、碱、氧化、高温条件下,均产生杂质B,且各破坏溶液色谱图中,主峰峰纯度及主峰与相邻杂质的分离度均符合要求.
2.2.3 重复性
取供试品溶液6份,分别取10 μL注入液相色谱仪测定杂质含量.结果显示,杂质均未检出,主峰峰面积的RSD为0.4%,小于1%,表明本方法重复性较好.
2.2.4 检测限和定量限
取左旋米那普仑有关物质A~E溶液,逐步稀释后进样10 μL,以信噪S/N约为3和10时的量作为检测限和定量限.结果表明,杂质A~E检测限分别为0.1720、0.5270、0.8667、0.8892、0.0631 μg/mL;杂质A~E定量限分别为0.5160、 0.8783、2.600、2.668、0.1683 μg/mL.
2.2.5 溶液稳定性
取“2.2.3”项下一份供试品溶液,于常温放置1、3、5、6、8、9、16、26 h后测定.结果表明,无新增杂质检出,主峰峰面积RSD为0.3%,均小于1%,说明常温下,供试品溶液在26 h内稳定.另取混合对照溶液,于常温放置1、2、3、4、5、8、12、16、21、29 h后测定.结果表明,杂质A~E的峰面积RSD分别为2.1%、1.7%、3.7%、3.1%、0.25%,均小于4%.结果表明,常温下,对照溶液在29 h稳定.
2.2.6 耐用性
取盐酸左旋米那普仑有关物质A~E与盐酸左旋米那普仑标准品适量,配制每1 mL含有关物质A~E各5 μg和盐酸左旋米那普仑5 mg的混和溶液,作为耐用性考察溶液.调整色谱条件的流速±0.1 mL/min、柱温±3 ℃或更换色谱柱,取该溶液进样.结果表明,在不同条件下,按峰面积百分比法计算各成分的量几乎不变,杂质与杂质之间以及杂质与左旋米那普仑之间分离度均大于1.5,表明本方法耐用性良好.
2.2.7 线性关系
取盐酸左旋米那普仑有关物质A~E与盐酸左旋米那普仑标准品适量,配制每1 mL含有关物质A~E与盐酸左旋米那普仑各50 μg/mL混和溶液作为线性储备液,并分别精密量取0.5 mL、0.8 mL、1.0 mL、1.2 mL、1.5 mL,置于不同的10 mL量瓶中,分别用10%乙腈(稀释剂)定容,作为线性关系考察样品溶液.取以上溶液各10 μL进样,以浓度和峰面积进行线性拟合.计算结果显示:杂质A,在2.58~7.74 μg/mL浓度范围内,回归方程为Y=0.4731X-0.0353(R2=0.9996);杂质B,在2.635~7.905 μg/mL浓度范围内,回归方程为Y=0.3629X+0.0102(R2=0.9965);杂质C,在2.6~7.8 μg/mL浓度范围内,回归方程为Y=0.1786X-0.0438(R2=0.9895);杂质D,在2.6675~8.0025 μg/mL浓度范围内,回归方程为Y=0.1322X-0.054(R2=0.9982);杂质E,在2.525~7.575 μg/mL浓度范围内,回归方程为Y=1.4049X-0.1972(R2=0.998);盐酸左旋米那普仑,在2.500~7.500 μg/mL浓度范围内,回归方程为Y=0.3849X+0.0949(R2=0.9963).结果表明,在相应浓度范围内,有关物质A~E与盐酸左旋米那普仑的进样浓度与峰面积线性关系良好.
2.2.8 准确度
取供试品适量,加10%乙腈溶解并稀释制成每1 mL约含50 mg的溶液,摇匀,作为准确度储备液,取储备液1 mL至10 mL量瓶中,分别精密加入0.8、1.0、1.2 mL“2.2.7”项下线性储备液(每个浓度平行制备3份),用10%乙腈稀释至刻度,摇匀,作为准确度供试液.取准确度供试液及“2.2.1”项下系统适用性溶液各10 μL进样,供试品各杂质含量均按均未检出计算,以杂质的测定量和加入量计算回收率.计算结果显示:杂质A的回收率范围为95.78%~97.88%,平均回收率为96.6%,RSD为0.69%;杂质B的回收率范围为97.80%~106.87%,平均回收率为101.5%,RSD为2.8%;杂质C的回收率范围为91.61%~109.76%,平均回收率为97.33%,RSD为6.5%;杂质D的回收率范围为90.13%~105.99%,平均回收率为94.9%,RSD为5.7%;杂质E的回收率范围为98.08%~101.30%,平均回收率为99.6%,RSD为1.0%.结果表明,各杂质回收率范围均在80%~120%,RSD均≤10%,本方法准确度良好.
3 讨 论
3.1 降解杂质结构的确认
实验显示,盐酸左旋米那普仑碱破坏供试液中检出2个较明显的杂质,即杂质1和杂质3(见图4).通过对该溶液进行LC-MS结构解析,初步确定两降解杂质的分子量分别为192与174,通过对盐酸左旋米那普仑(分子量247)结构式进行拆分,本研究确定2个杂质的结构式为图1中的杂质B和杂质D.
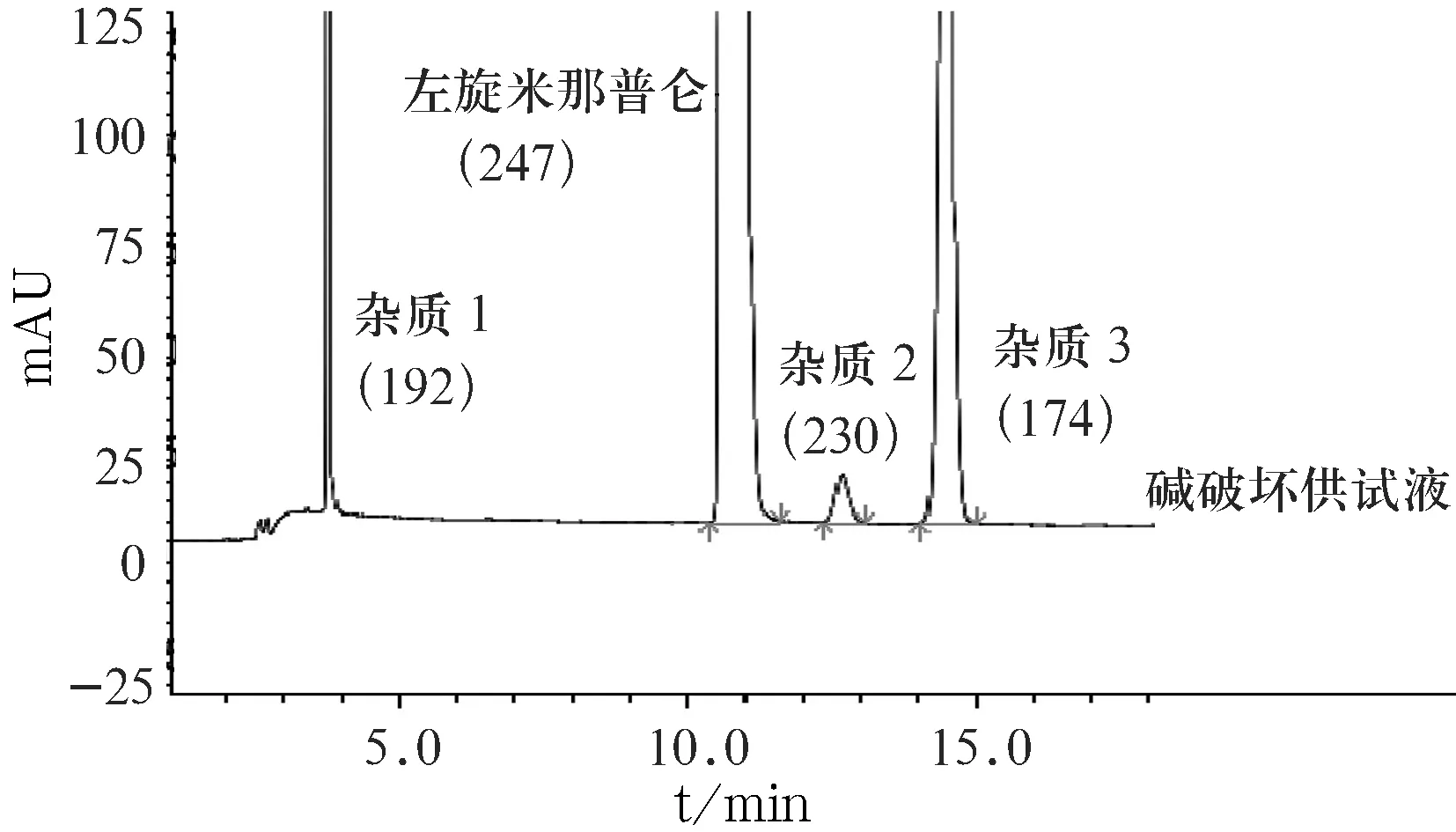
图4供试液碱破坏液相色谱图
3.2 紫外吸收光谱检测
“2.2.1”项下杂质A~E和盐酸左旋米那普仑的定位溶液各主峰光谱图(见图5).结果显示,除杂质E在190 nm、220 nm处有较明显的吸收峰外,其他杂质和盐酸左旋米那普仑除在190 nm外,无明显的吸收峰.为了避开末端吸收(190 nm)的干扰,同时确保各杂质和主成分均有较强的吸收,本研究选择220 nm作为检测波长.

图5杂质与米那普仑光谱图
3.3 水相添加剂的筛选
相关研究显示,杂质D极性很强,需要很低比例的有机相,而杂质E极性太差,需要较高比例的有机相.本实验发现,当以纯水为水相时,主峰拖尾严重,而以磷酸盐为水相时,杂质E附近会出现明显的鬼峰(高比例乙腈与磷酸盐混合,出现盐析现象所致),当以0.05%TFA水溶液为水相时,不仅主峰和杂质峰峰型良好,且杂质E附近基线平稳.
4 结 论
实验的结果表明,本研究建立的分析方法,各成分分离良好,同时可兼顾极性小的工艺杂质和极性大的降解杂质,具有简便快速、准确灵敏、重复性好的特点,完全适用于盐酸左旋米那普仑有关物质测定.
猜你喜欢
科学与财富(2021年36期)2021-05-10
中学生数理化·高一版(2021年2期)2021-03-19
中学生数理化·高一版(2021年2期)2021-03-19
中学生数理化(高中版.高二数学)(2019年6期)2019-06-24
布达拉(2019年3期)2019-06-11
中国盐业(2018年20期)2019-01-14
中国当代医药(2015年33期)2015-03-01
郑州大学学报(医学版)(2015年2期)2015-02-27
中华皮肤科杂志(2014年4期)2014-12-19
中华皮肤科杂志(2014年3期)2014-12-19
