
乙酰苯胺纯度标准物质的定值及不确定度评定
2020-01-09 00:37孟娇然夏娃段嫚雷齐钰余金汭上海市计量测试技术研究院
上海计量测试 2019年6期
孟娇然 夏娃 段嫚雷 齐钰 余金汭 / 上海市计量测试技术研究院
0 引言
乙酰苯胺作为有机工业合成重要的中间体,是磺胺类药物、染料、樟脑等的原料,同时也是橡胶硫化过程的促进剂[1],被广泛应用于化工、纺织、制药等领域。随着乙酰苯胺的应用越来越广泛,乙酰苯胺纯度标准物质的需求也日益提升。该标准物质不仅可用于元素分析仪校准,还可用于熔点测定的量值传递等。
有机纯物质纯度的定值方法主要有质量平衡法、定量核磁法、元素分析法、滴定法、凝固点下降法和差示扫描量热法(DSC)等[2]。其中质量平衡法也称为杂质扣除法,是通过测量其中所有杂质的含量,从100%中减去杂质的质量分数,获得主体成分的质量分数即纯度值的方法。质量平衡法能够克服不同物质理化性质造成响应值差异的缺点,通过准确测定各杂质含量,从而得到样品的纯度[3]。DSC法则是一种热分析技术,根据物质的熔点与纯度之间的关系,基于范德霍夫(Van’t Hoff)方程计算物质的纯度。该方法适用于加热过程中不发生热分解、不与外界产生热效应的有机纯物质的纯度分析[4]。
本研究采用区域熔融法对乙酰苯胺进行提纯,采用质量平衡法和DSC法两种方法对乙酰苯胺纯度标准物质进行纯度定值,并评定其不确定度。
1 实验部分
1.1 主要试剂和仪器
乙酰苯胺(分析纯,国药集团);甲醇(色谱纯,Merck)。
1100型高效液相色谱仪(美国Agilent公司);Diamond型差示扫描量热仪(美国Perkin-Elmer公司);931型卡尔·费休水分测定仪(瑞士Metrohm公司);XS205型电子天平(瑞士Mettler-Toledo公司)。
1.2 乙酰苯胺纯度标准物质的制备
乙酰苯胺的提纯以分析纯乙酰苯胺作为原料,采用区域熔融法在150 ℃下,以45 mm/h移动速率对样品进行反复区域熔融,得到白色结晶状提纯产物,经研磨并充分混匀后得到乙酰苯胺纯度标准物质。
1.3 定值分析
采用质量平衡法和DSC法对乙酰苯胺纯度标准物质进行定值。
1.3.1 质量平衡法
乙酰苯胺纯度标准物质中的杂质分别采用高效液相色谱法,卡尔·费休库仑滴定法和炽灼残渣法进行定值检测。
1.3.1.1 高效液相色谱法检测条件
色谱柱:Waters C18 填充柱(4.6 mm×150 mm,3.5 μm);流动相:甲醇 /水(50/50);流量:1 mL/min;检测波长:230 nm;进样体积:20 μL;乙酰苯胺浓度:10 mg/mL。
1.3.1.2 水分含量测定
水分含量采用卡尔·费休库仑滴定法测定,每次测量称取乙酰苯胺纯度标准物质2 g (称量准确至0.01 mg),样品平行测量6次。
1.3.1.3 不挥发性组分测定
不挥发性组分的测定参照2015版《中国药典》的炽灼残渣法[5],每次称取乙酰苯胺纯度标准物质 5~6 g(称量准确至 0.01 mg)置于已恒重的坩埚内,炽灼至完全炭化,经硫酸酸化处理,待硫酸蒸汽除尽后,再置于马弗炉内以800 ℃炽灼使其完全灰化,恒重后计算不挥发组分的含量。样品平行测量6次。
1.3.2 差示扫描量热法检测条件
准确称取乙酰苯胺纯度标准物质1.0 mg(称量准确至0.01 mg)至铝坩埚内,密封压盖,置于量热池内,采用氮气作为保护气,流量为20 mL/min。以20 ℃/min的速率升温至100 ℃,再以0.7 ℃/min的速率升温至115 ℃,重复测定6次。
1.4 均匀性检验
乙酰苯胺纯度标准物质封装在棕色玻璃瓶内,每瓶2 g。从中随机抽取15瓶,采用1.3.2所述DSC方法对该15瓶标准物质进行纯度检测,每瓶重复测定3次。
1.5 稳定性检验
在制备时间后0、1、3、6、9、12个月六个时间点随机抽取3瓶乙酰苯胺纯度标准物质样品,采用1.3.2所述DSC方法对其纯度进行跟踪测量。
2 实验结果与讨论
根据JJF 1343-2012《标准物质定值的通用原则及统计学原理》[6],标准物质可采用两种或两种以上测量原理不同、具有明确溯源性和不确定度的方法对其进行定值。在本研究中对乙酰苯胺纯度标准物质采用质量平衡法和DSC法定值,可以避免一种方法可能带来的技术缺陷。
2.1 质量平衡法定值
质量平衡法定值过程中,乙酰苯胺的纯度采用高效液相色谱归一化法结合含水量检测、不挥发性组分检测进行确定。
乙酰苯胺中的有机杂质采用高效液相色谱法进行分析检测。乙酰苯胺标准物质中最可能存在的有机杂质为苯胺,根据1.3.1.1所示方法对乙酰苯胺标准物质进行分析,在该条件下乙酰苯胺和苯胺的保留时间分别为4.70 min和7.29 min。实验中未检测到苯胺以及其他杂质的色谱峰。
根据高效液相色谱、水分含量和不挥发性组分测试结果,乙酰苯胺标准物质采用质量平衡法测得的纯度值数据如表1所示。
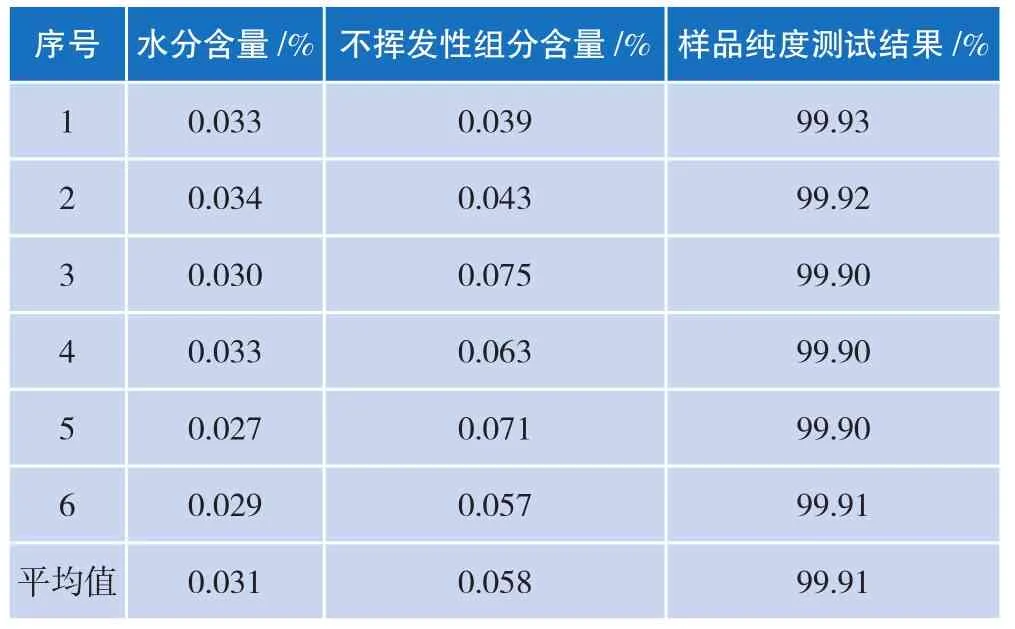
表1 乙酰苯胺采用质量平衡法的定值结果
2.2 DSC法定值
除质量平衡法外,参照GB/T 28723-2012《固体有机化学品纯度的测定差示扫描量热法》[7],采用差示扫描量热仪对乙酰苯胺标准物质的纯度进行检测。由于物质熔点下降与杂质含量的关系符合范德霍夫方程,因此可以通过测定物质的熔点计算其中的杂质含量,并由此得到物质的纯度,即纯度%=100%-杂质%。本实验中根据仪器的纯度计算软件直接计算得到相应的纯度数值,测试结果如表2所示。

表2 乙酰苯胺采用DSC法纯度的定值结果
2.3 定值方法等准确度、一致性检验
由于乙酰苯胺纯度标准物质是通过质量平衡法和DSC法两种不同原理的方法进行定值,因此需对这两种定值方法的数据进行等准确度检验和平均值一致性检验。对表1和表2数据进行F检验和t检验。当α为0.05时,纯度定值数据的F值为0.47,满足f2α(5,5)= 0.14 <F<f1α(5,5)= 7.15 的要求,F检验通过即证明两种方法等准确度。另外,计算得到的t值也满足 |t| <t0.05,(10)= 2.23,t检验通过证明两种方法定值结果一致。由于定值结果同时通过等准确度检验和平均值一致性检验,因此最终纯度定值结果为两者的算术平均值,即
2.4 均匀性检验结果
采用DSC法对乙酰苯胺纯度标准物质随机抽取的15个单元进行均匀性检验,数据分析采用方差分析法进行,通过组间方差和组内方差的比较来判断各组测量值之间有无系统性差异,即进行F检验,数据如表3所示。

表3 乙酰苯胺的均匀性检验数据
计算得组内方差和= 0.000 471,组间方差和= 0.000 213,经统计分析两者之比F= 2.21,小于临界值F0.01(14,30)= 2.74,符合 JJF 1343-2012 的要求,证明标准物质的均匀性良好。
2.5 稳定性检验结果
标准物质的稳定性除了受自身性质的影响外,同样会受其制备过程、储存容器、外部储存条件等影响。为保证乙酰苯胺标准物质的稳定,将乙酰苯胺纯度标准物质分装于棕色玻璃瓶内存放于阴凉干燥处,同时选取6个时间点做稳定性测试。采用直线拟合法和t检验对乙酰苯胺纯度标准物质的稳定性检验结果进行评价,数据如表4所示。
以各时间点纯度测量值对时间拟合直线,计算得直线斜率的绝对值 |b1| = 0.002 71,该数据小于临界值t(0.95,4)·s(b1) = 0.003 70,结果表明直线的斜率不显著,证明乙酰苯胺纯度标准物质在12个月内无明显的上升或下降趋势。

表4 乙酰苯胺的稳定性检验数据
3 乙酰苯胺的不确定度评定
根据乙酰苯胺纯度标准物质定值过程中的影响因素,对定值结果的不确定度进行评定,主要包括标准物质纯度定值过程中引入的不确定度,均匀性引入的不确定度和稳定性引入的不确定度。
3.1 标准物质纯度定值过程中引入的不确定度(uchar)
由于乙酰苯胺纯度标准物质由质量平衡法和DSC法两种方法定值,因此标准物质定值过程中引入的不确定度同时来源此两种定值方法引入的不确定度。
3.1.1 质量平衡法引入的不确定度
基于质量平衡法的纯度值的不确定来源分别为苯胺含量引入的u(Xani),水分含量引入的u(XW)和不挥发性组分含量引入的u(XNV),且各分量互不相关。
由于高效液相色谱法未检出苯胺杂质,因此苯胺含量引入的不确定度主要由仪器对苯胺的检出限0.005 mg/L 引入,u(Xani)为 0.05%。
水分含量测定引入的不确定度u(XW)为0.001 44%。
不挥发性组分含量测定的不确定度u(XNV)为0.009%。
根据上述数据,采用质量平衡法计算得到的纯度值的标准不确定度为

3.1.2 DSC法引入的不确定度
基于DSC法的纯度值的不确定度u(P2)即为杂质含量的不确定u(xB)。
杂质含量的不确定度u(xB)来源主要包括测量重复性,仪器温度示值误差和仪器热量示值误差引入的不确定度,且各分量互不相关。
根据表2数据可知,由重复性引入的相对标准不确定度为urel为13.88%
差示扫描量热仪温度引入的相对不确定度为urel(T)为1.01%
差示扫描量热仪热量引入的相对不确定度为urel(H)为2.89%
根据上述数据,采用DSC法计算得到的纯度值的标准不确定度为
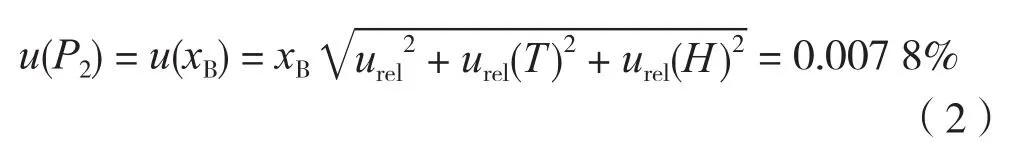
3.1.3 纯度定值不确定度
由于2.3中已证明质量平衡法和DSC法等准确度且平均值的结果一致,因此纯度定值的标准不确定度为

3.2 均匀性引入的不确定度
根据乙酰苯胺的均匀性检验数据(表3),由均匀性导致的不确定度分量为
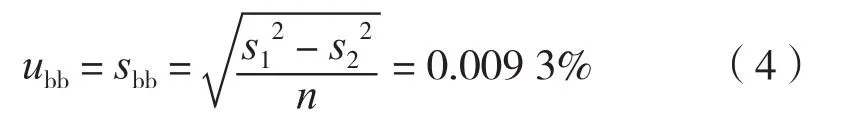
3.3 稳定性引入的不确定度
根据乙酰苯胺的稳定性分析数据(表4),由稳定性导致的不确定度分量为

3.4 合成不确定度和扩展不确定度
综上,乙酰苯胺纯度标准物质的不确定度为定值过程引入的不确定度uchar、均匀性引入的不确定度ubb和稳定性引入的不确定度us三个分量的合成,即:
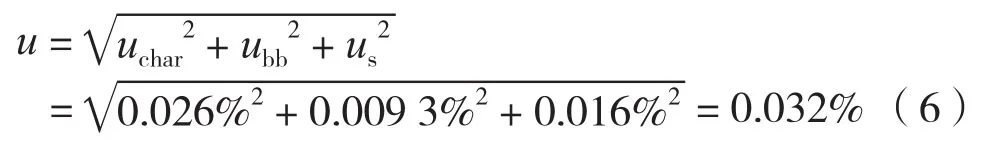
取包含因子k= 2,置信度水平约为95%,扩展不确定度为U= 0.064%≈0.1%。因此,该批次乙酰苯胺纯度标准物质的定值结果为99.9%±0.1%(k= 2)。
4 结语
采用质量平衡法和DSC法对乙酰苯胺纯度标准物质进行纯度定值,同时对其不确定度进行评定,得到乙酰苯胺标准物质的纯度结果为99.9%,扩展不确定度为U= 0.1%(k= 2)。通过F检验和t检验证明了该标准物质具有良好的均匀性和稳定性,有效期限为12个月。
猜你喜欢
能源化工(2022年1期)2023-01-14
分子催化(2022年1期)2022-11-02
中学生数理化(高中版.高二数学)(2022年1期)2022-04-26
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
新世纪智能(教师)(2021年2期)2021-11-05
电子制作(2018年10期)2018-08-04
电子制作(2018年12期)2018-08-01
云南中医学院学报(2015年2期)2015-07-31
质谱学报(2015年5期)2015-03-01
