
三种国标方法测定酒中甜蜜素的比较与方法改进
2020-01-08 05:07闫吉昌马长海于明明辛若竹
酿酒科技 2019年12期
闫吉昌,马长海,于明明,辛若竹
(1.白城市产品质量检验所,吉林白城 137000;2.梅河口市食品药品检验检测中心,吉林梅河口 135000)
甜蜜素虽然作为一种甜味剂广泛应用于各类食品中,但长期过量摄入也会损害健康,所以国家食品安全标准[1]严格规定了其使用范围和使用限量。酒类产品按生产工艺和产品特性分为蒸馏酒、发酵酒和配制酒[2]。甜蜜素常作为甜味剂应用于配制酒中,这是国家食品添加剂使用标准允许的。而不法商贩为改善酒品口感将甜蜜素应用于蒸馏酒和发酵酒中,这却是国家严格禁止的。所以为保证酒类产品的品质,对酒中甜蜜素监测是十分必要的。目前,甜蜜素测定的常用方法有气相色谱法[3-4]、液相色谱法[5-6]、液相色谱-质谱/质谱法[7-9]。GB 5009.97—2016甜蜜素测定的国家标准检测方法[10]中规定了不同种类产品与其相应适用的分析方法,其中气相色谱法不适用于酒类产品测定,液相色谱法适用于配制酒检测,液相色谱-质谱/质谱法适用于蒸馏酒和发酵酒测定。本文通过比较气相色谱法、液相色谱法、液相色谱-质谱/质谱法3种国标方法测定酒中甜蜜素含量,发现国标气相色谱法不能将酒中的干扰物分离,常出现甜蜜素假阳性结果,这与其规定的适用范围相吻合;液相色谱法虽无基质干扰现象,其检出限满足配制酒的检测要求,但样品衍生化时会产生有害物质,对操作者有潜在的危害;液相色谱-质谱/质谱法虽是检测酒中甜蜜素最佳方法,但其设备比较昂贵,应用范围受限。为了找到实用性更好且对操作者无潜在危害的酒中甜蜜素的检测方法,对国标气相色谱法进行了改进,通过优化色谱条件,使改进后的气相色谱法能有效地消除基质干扰,检出限、回收率及精密度均能满足配制酒日常检测要求,且方法技术参数均优于液相色谱法,扩展了国标气相色谱法的适用范围。同时对国标液相色谱-质谱/质谱法进行了优化,优化后的方法检出限为0.01 mg/kg,低于国标液相色谱-质谱/质谱法,能满足酒中微量甜蜜素的检测要求。
1 材料与方法
1.1 材料、试剂及仪器
酒样:市售白酒。
试剂及耗材:甜蜜素标准物质(纯度99.1 %,Dr.Ehrenstorfer);色谱纯甲醇、乙腈(Fisher Scientific);色谱纯正庚烷(CNW);色谱纯甲酸(CNW);氢氧化钠溶液(40 g/L);硫酸溶液(200 g/L);亚硝酸钠溶液(50 g/L);氯化钠;硫酸溶液(1+1);次氯酸钠溶液(有效氯含量50 g/L以上);碳酸氢钠溶液(50 g/L)。
仪器及设备:GC2010plus气相色谱仪(FID检测器,日本岛津公司);HP-5MS色谱柱(30 m×0.25 mm×0.25 μm);Agilent 1260高效液相色谱仪(DAD检测器,美国安捷伦公司);Agilent Poroshell 120 EC-C18色谱柱(2.7 μm,3.0×150 mm);AB SCIEX API4000+超高效液相色谱/串联质谱联用仪(ESI离子源,美国AB SCIEX公司);Kinetex® 2.6 μm F5 100 Å色谱柱(50×3.0 mm);涡旋混合器(美国talboys);KH20R-Ⅱ高速冷冻离心机(湖南凯达);DK-98-1型电热恒温水浴锅(天津市泰斯特仪器有限公司)。
1.2 样品处理
1.2.1 气相色谱法和液相色谱法样品溶液制备
称取酒样25.0 g于烧杯中,用氢氧化钠溶液(40 g/L)调至pH7~8,60 ℃水浴加热30 min以除去酒精,放冷,用水定容至50 mL备用。
1.2.2 气相色谱法衍生化
准确移取制备好的酒样溶液10.0 mL于50 mL带盖离心管中,置冰浴中5 min后,准确加入5.00 mL正庚烷,加50 g/L亚硝酸钠溶液2.5 mL,加200 g/L硫酸溶液2.5 mL,盖紧离心管盖,摇匀,在冰浴中放置30 min,期间振摇3~5次;加入2.5 g氯化钠,盖上盖后涡旋1 min,低温3000 r/min离心10 min分层后,取上清液放置1~4 ℃冰箱冷藏保存以备进样。
1.2.3 液相色谱法衍生化
准确移取制备好的酒样溶液10.0 mL于50 mL带盖离心管中,加入2.0 mL硫酸溶液(1+1),5.0 mL正庚烷和1.0 mL次氯酸钠溶液(有效氯含量50 g/L以上),剧烈振荡1 min,静置分层,除去水层后在正庚烷层中加入25 mL碳酸氢钠溶液(50 g/L),振荡1 min。静置取上层有机相经0.45 μm微孔有机相滤膜过滤,滤液进样备用。
1.2.4 液相色谱-质谱/质谱法样品前处理
称取酒样1.0 g于50 mL烧杯中,加水1.0 mL,于60 ℃水浴上加热30 min,残渣全部转移至10 mL容量瓶中,用水定容并摇匀,经0.22 μm水相微孔滤膜过滤后备用。
1.3 标准溶液制备
1.3.1 标准溶液配制(1.00 mg/mL)
精确称取0.1124 g环己基氨基磺酸钠标准品,用水溶解并定容至100 mL,混匀,此溶液1.00 mL相当于环己基氨基磺酸1.00 mg。
1.3.2 标准工作溶液系列1
准确吸取1.00 mg/mL标准溶液0.5 mL、1.00 mL、2.50 mL、5.00 mL、10.0 mL、25.0 mL于50 mL容量瓶,加水定容,混匀。配制的标准溶液系列浓度分别为10 μg/mL、20 μg/mL、50 μg/mL、100 μg/mL、200 μg/mL、500 μg/mL。准确移取标准系列溶液10.0 mL分别按1.2.2衍生化于气相色谱分析,再分别按1.2.3衍生化于用于液相色谱分析。
1.3.3 标准工作溶液系列2
准确吸取10 μg/mL标准工作溶液0.01 mL、0.05 mL、0.1 mL、0.5 mL、1.0 mL于10 mL容量瓶,加水定容,混匀。配成标准工作溶液系列浓度分别为0.01 μg/mL、0.05 μg/mL、0.1 μg/mL、0.5 μg/mL、1.0 μg/mL。用于液相色谱-质谱/质谱分析。
1.4 色谱条件
1.4.1 气相色谱条件
HP-5MS石英毛细管柱(30m×0.25mm×0.25μm),进样口温度230 ℃,检测器温度260 ℃。柱流量1.00 mL/min,氢气流速:40.0 mL/min,空气流速:400.0 mL/min。分流进样,分流比1∶10,进样量1 μL。原国标法柱温升温程序55 ℃保持3 min,10 ℃/min升至90 ℃保持0.5 min,20 ℃/min升至200 ℃保持3 min。优化后的柱温升温程序55 ℃保持3 min,1 ℃/min升至60 ℃保持2 min,20 ℃/min升至200 ℃保持3 min。
1.4.2 液相色谱条件
Poroshell 120 EC-C18色谱柱2.7 μm,3.0×150 mm,流动相:乙腈+水(70+30),流速0.3 mL/min;检测波长:314 nm;柱温40 ℃;进样量5 μL。
1.4.3 优化的液相色谱-质谱/质谱条件
1.4.3.1 色谱条件:Kinetex®2.6 μm F5 100 Å色谱柱50×3.0 mm,流动相:A泵为0.1%(v/v)甲酸水,B泵为甲醇,梯度洗脱程序B泵:0 min 3%;1 min,3 %;1.1 min,15 %;5.0 min,40 %;5.1 min,95 %;7.0 min,95 %;7.1 min,3 %;10 min,3 %。流速0.4 mL/min;柱温40 ℃;进样量5 μL。
1.4.3.2 质谱条件
①负离子模式:
离子源:ESI;检测方式:-MRM;离子源参数:电喷雾电压(IS):-4500 V;雾化气压力(GS1):55 Psi;辅助气压力(GS2):60 Psi;气帘气压力(CUR):30 Psi;离子源温度(TEM):550 ℃;喷撞气(CAD):7 mL/min。监测离子对及电压参数:定量离子对Q1/Q3:178.0/79.9 m/z;驻留时间:200 ms;去簇电压(DP):-60 V;碰撞气能量(CE):-35 V;碰撞池入口电压(EP):-10 V;碰撞池出口电压(CXP):-15 V。
②正离子模式:
离子源:ESI;检测方式:+MRM;离子源参数:电喷雾电压(IS):5500 V;雾化气压力(GS1):55 Psi;辅助气压力(GS2):60 Psi;气帘气压力(CUR):30 Psi;离子源温度(TEM):550 ℃;喷撞气(CAD):7 mL/min。监测离子对及电压参数:定性离子对Q1/Q3:202.2/122.1m/z;驻留时间:500 ms;去簇电压(DP):50 V;碰撞气能量(CE):11.8 V;碰撞池入口电压(EP):10 V;碰撞池出口电压(CXP):13 V。
1.5 样品测定
1.5.1 气相色谱法
分别吸取1 μL经衍生化处理的标准系列各浓度溶液上清液,注入气相色谱仪中,可测得不同浓度被测物的响应值峰面积,以浓度为纵坐标,以环己醇亚硝酸酯和环己醇两峰面积之和为横坐标,绘制标准曲线。在完全相同的条件下进样1 μL经衍生化处理的试样待测液上清液,保留时间定性,测得峰面积,根据标准曲线计算样液中的甜蜜素浓度。
1.5.2 液相色谱法
分别吸取5 μL经衍生化处理的标准系列各浓度溶液上清液,注入液相色谱仪中,可测得不同浓度被测物的响应值峰面积,以浓度为横坐标,以环己基氨基磺酸钠衍生化产物N,N-二氯环己胺峰面为纵坐标,绘制标准曲线。在完全相同的条件下进样5 μL经衍生化处理的试样待测液上清液,保留时间定性,测得峰面积,根据标准曲线计算样液中的甜蜜素浓度。
1.5.3 液相色谱-质谱/质谱法
1.5.3.1 定量测定
将配制好的标准工作溶液系列2按照浓度由低到高的顺序进样测定,以环己基氨基磺酸钠定量离子的色谱峰面积对相应的浓度作图,得到标准曲线回归方程。将试样溶液注入液相色谱-质谱/质谱仪中,得到环己基氨基磺酸钠定量离子峰面积,根据标准曲线计算试样溶液中环己基氨基磺酸钠的浓度。
1.5.3.2 定性测定
在相同的试验条件下测定试样溶液,若试样溶液质量色谱图中环己基氨基磺酸钠的保留时间与标准溶液一致(变化范围在±2.5%以内),且试样定性离子的相对丰度与浓度相当的标准溶液中定性离子的相对丰度,其偏差不超过规定要求(相对丰度>50 %,允许±20 %偏差;相对丰度>20 %~50%,允许±25%偏差;相对丰度>10%~20%,允许±30%偏差;相对丰度≤10%,允许±50%偏差),则可判定样品中存在环己基氨基磺酸钠。
2 结果与分析
2.1 液相色谱-质谱/质谱法优化
2.1.1 前处理方法改进
取酒样1.0 g,加1 mL水,于60 ℃水浴上加热30 min,残渣全部转移并定容至10 mL。这样能将酒中的挥发成分彻底去除,降低本底干扰,提高甜蜜素质谱响应信号。
2.1.2 色谱条件优化
由于甜蜜素的负离子对178.0/79.9 m/z的响应信号特强,而正离子对202.2/122.1 m/z响应信号相对较弱,针对此特征,本文采用0.1%(v/v)甲酸水-甲醇体系为流动相,提高甜蜜素的正离子对的响应值,同时与国标法相比,流动相未引入铵盐,降低了质谱系统损坏的风险。
2.1.3 方法的校准曲线和检出限
按照本文液相色谱串联质谱法优化的色谱条件,对甜蜜素标准工作溶液系列2进行测定,绘制标准曲线,结果表明,浓度在0.01~0.05 μg/mL范围内有良好的线性关系,回归方程y=4479180x+10749和相关系数R=0.9999;甜蜜素的负离子质谱响应信号远大于正离子响应信号,以正离子模式信噪比为响应,计算方法检出限更具有实际意义。图1为标准曲线第一个浓度点0.01 μg/mL测得正离子模式下信噪比图,以3倍信噪比(S/N=3)计算方法的检出限为0.01 mg/kg,以10倍信噪比(S/N=10)计算方法的定量限为0.03 mg/kg。说明该方法的检出限远远低于国标方法的检出限,能满足酒中微量甜蜜素的检测要求。

图1 正离子模型下的信噪比图
2.1.4 方法的精密度和加标回收率
选取一市售白酒样品,按3个浓度水平加入甜蜜素标准溶液,制备成加标样品,分别对白酒样品、白酒加标样品进行测定,对每个添加水平重复测定6次,计算加标回收率和相对标准偏差,数据见表1。甜蜜素的MRM色谱图见图2。结果表明,酒样中甜蜜素的加标回收率在90%~108%之间,相对标准偏差在0.69%~1.78%的范围内,说明该方法准确、稳定、可靠。
2.2 液相色谱法分析结果
2.2.1 酒样本底干扰试验
将上述经液相色谱-质谱/质谱法测定无甜蜜素的白酒样品,添加甜蜜素标准溶液制备成加标样品(加标浓度100 mg/kg),按照液相色谱法分别对白酒样品、白酒加标样品及甜蜜素标液(100 μg/mL)进行测定,得到白酒样品、白酒加标样品及甜蜜素标液的色谱图。对比白酒样品色谱图(图3a)和白酒加标样品色谱图(图3b)及甜蜜素标液色谱图(图3c)可以看出,白酒样品在甜蜜素出峰时间点上未出现色谱峰,白酒加标样品和甜蜜素标液均在甜蜜素出峰时间点上出现了色谱峰,说明液相色谱法测定白酒中甜蜜素无本底干扰。
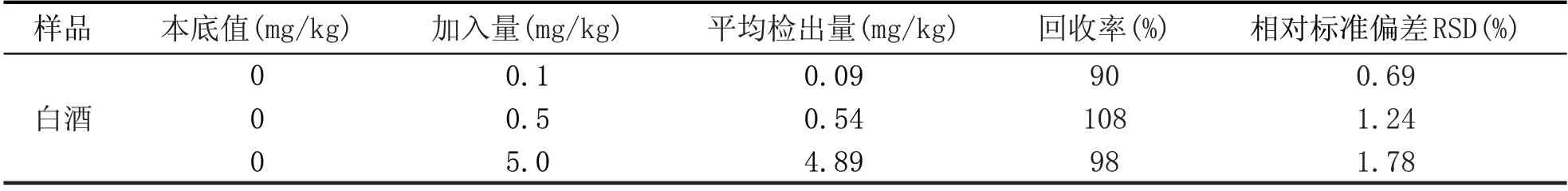
表1 液相色谱-质谱/质谱法回收率及精密度测定结果(n=6)

图2 白酒样品、白酒加标样品、甜蜜素标液的多反应监测(MRM)色谱图
2.2.2 液相色谱法技术参数考察
按照本文液相色谱法,对甜蜜素标准工作溶液系列1进行测定,绘制标准曲线;以3倍信噪比(S/N=3)计算方法的检出限,以10倍信噪比(S/N=10)计算方法的定量限。同时将2.2.1中测得的加标样品计算的回收率、精密度均列入表2中。由表2可知,液相色谱法的方法检出限虽然不能满足国家标准对白酒(蒸馏酒)的检测要求,但完全满足配制酒的检测要求。然而液相色谱法存在不足之处,样品衍生化时会产生有害物质,对操作者有潜在的危害。
2.3 气相色谱法分析结果
2.3.1 国标气相色谱法酒样本底干扰试验

表2 液相色谱法技术参数
将上述经液相色谱-质谱/质谱法测定无甜蜜素的白酒样品,添加甜蜜素标准溶液制备成加标样品(加标浓度20 mg/kg),按照国标气相色谱法分别对白酒样品、白酒加标样品及甜蜜素标液(100 μg/mL)进行测定,得到白酒样品、白酒加标样品及甜蜜素标液的色谱图。对比白酒样品色谱图(图4a)和白酒加标样品色谱图(图4b)及甜蜜素标液色谱图(图4c)可以看出,白酒样品、白酒加标样品和甜蜜素标液均在甜蜜素出峰时间点上出现了色谱峰,说明国标气相色谱法测定白酒中甜蜜素有本底干扰。
2.3.2 改进气相色谱法酒样本底干扰试验
按照改进气相色谱法分别对上述白酒样品、白酒加标样品(加标浓度20 mg/kg)及甜蜜素标液(100 μg/mL)进行测定,得到白酒样品、白酒加标样品及甜蜜素标液的色谱图。对比白酒样品色谱图(图5a)和白酒加标样品色谱图(图5b)及甜蜜素标液色谱图(图5c)可以看出,不含甜蜜素的白酒样品在甜蜜素出峰时间点上未出色谱峰,白酒加标样品和甜蜜素标液均在甜蜜素出峰时间点上出现了色谱峰,说明改进气相色谱法测定白酒中甜蜜素无本底干扰。
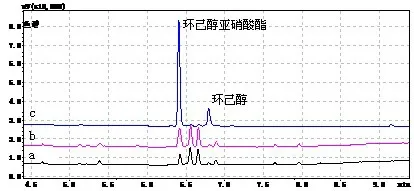
图4 白酒样品、白酒加标样品、甜蜜素标液国标法气相色谱图比较

图5 白酒样品、白酒加标样品、甜蜜素标液改进法气相色谱图比较
2.3.3 改进气相色谱法技术参数考察
按照本文改进气相色谱法,对甜蜜素标准工作溶液系列1进行测定,绘制标准曲线;以3倍信噪比(S/N=3)计算方法的检出限,以10倍信噪比(S/N=10)计算方法的定量限。同时将2.3.2中测得的加标样品计算的回收率、精密度均列入表3中。由表3可知,改进气相色谱法的方法检出限虽然不能满足国家标准对白酒(蒸馏酒)的检测要求,但完全满足配制酒的检测要求,而且方法技术参数均略优于液相色谱法,且对操作者无潜危害,实用性更好。
3 结论
通过比较分析3种国标方法测定白酒中甜蜜素含量,得出以下结论:优化的液相色谱-质谱/质谱法检出限更低,方法准确、可靠,更适合于白酒(蒸馏酒)中微量甜蜜素的测定;液相色谱法检出限满足配制酒的检测要求,但存在不足之处是样品衍生化时会产生有害物质,对操作者有潜在的危害;国标气相色谱法不能将酒中的干扰物分离,常出现甜蜜素假阳性结果,改进的气相色谱法能有效地消除基质干扰,其检出限、回收率及精密度均能满足配制酒的检测要求,而且方法技术参数均优于液相色谱法,且对操作者无潜危害,实用性更好。
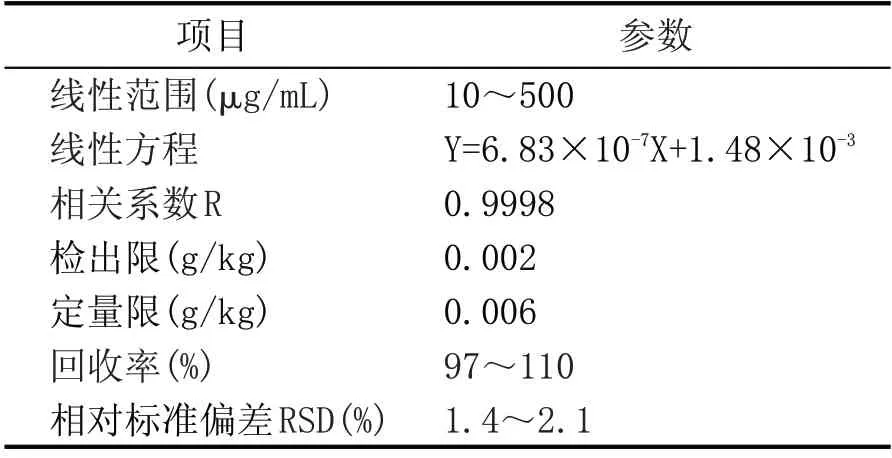
表3 改进气相色谱法技术参数
猜你喜欢
当代水产(2022年4期)2022-06-05
商品与质量(2021年43期)2022-01-18
口腔护理用品工业(2021年4期)2021-11-02
陶瓷学报(2021年4期)2021-10-14
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
