
槐猪与杜洛克猪背部皮下脂肪转录组分析
2020-01-04 07:18叶鼎承杨建敏甘乾福刘景
安徽农业科学 2020年24期
叶鼎承 杨建敏 甘乾福 刘景


摘要 [目的]了解槐猪和杜洛克猪背部皮下脂肪组织脂肪沉积的分子机制差异。[方法]对槐猪和杜洛克猪背部皮下脂肪组织进行转录组测序,并进行生物信息学分析。[结果]槐猪和杜洛克猪分别获得37 858 968和37 545 394条干净读段(clean reads)。槐猪和杜洛克猪分别预测到9 612和9 457可变剪接事件;两猪种间共有709个差异表达基因,与杜洛克猪相比,槐猪有395个上调基因和314个下调基因。差异表达基因共归类到46个GO条目中,其中细胞组分11个、分子功能12个及生物学过程23个;显著富集的KEGG通路共计9个,其中含有与调节脂类代谢显著相关通路,如胰岛素信号通路和PPAR信号通路等。[结论]该研究可为今后研究脂肪沉积的遗传机理提供科学依据。
关键词 槐猪;杜洛克猪;背部皮下脂肪组织;差异表达;转录组测序
中图分类号 S828;S813文献标识码 A
文章编号 0517-6611(2020)24-0099-03
doi:10.3969/j.issn.0517-6611.2020.24.027
开放科学(资源服务)标识码(OSID):
Transcriptome Analysis of Back Subcutaneous Fat of Huai Pig and Duroc Pig
YE Dingcheng1,YANG Jianmin2,GAN Qianfu3 et al (1.Institute of Animal Husbandry and Veterinary Medicine,Fujian Academy of Agricultural Sciences,Fuzhou,Fujian 350013;2.Animal Husbandry and Veterinary Station in Houyu Township of Changle District in Fuzhou City,Fuzhou,Fujian 350200;3.College of Animal Sciences,Fujian Agriculture and Forestry University,Fuzhou,Fujian 350002)
Abstract [Objective]To understand the molecular mechanism difference of fat deposition in the subcutaneous adipose tissue between Huai Pig and Duroc Pig.[Method]Transcriptome sequencing of subcutaneous adipose tissue in Huai Pig and Duroc Pig were performed and bioinformatic analysis was made.[Result]37 858 968 and 37 545 394 clean reads were obtained from Huai Pig and Duroc Pig ,respectively.9 612 and 9 457 alternative splicing events were predicted in Huai Pig and Duroc Pigrespectively. There were 709 differentially expressed genes (DEGs) in these two pig breeds.Compared with Duroc Pig,there were 395 upregulated genes and 314 downregulated genes in Huai Pig. The DEGs were classified into 46 gene ontology (GO) entries,including 11 cell components,12 molecular functions and 23 biological processes.A total of 9 pathways had enriched KEGG,which contained pathways significantly correlated with regulation of lipid metabolism,such as insulin signaling pathway and PPAR signaling pathway.[Conclusion]This study can provide scientific basis for the future study of the genetic mechanism of fat deposition.
Key words Huai Pig;Duroc Pig;Back subcutaneous adipose tissue;Differential expression;Transcriptome sequencing
脂肪沉積性状是猪主要的经济性状之一,且与经济价值紧密联系。槐猪(Huai Pig)是福建省特色猪品种,早熟易肥,肉质优良,抗逆性强,但生长速度慢,瘦肉率低,是典型的脂肪型猪种,而西方现代猪种杜洛克猪(Duroc Pig)生长速度快,瘦肉率高,但肉质和抗逆性相对较差,是典型的瘦肉型猪种。为了解槐猪和杜洛克猪皮下脂肪组织脂肪沉积的分子机制差异,笔者对这两个猪种背部皮下脂肪进行了转录组测序,并进行了生物信息学分析。
1 材料与方法
1.1 试验材料与测序
选择在相近饲养条件下同期饲养的190日龄槐猪和杜洛克猪各3头,屠宰后分别取倒数第3~4根肋骨之间的背部皮下脂肪组织,置于液氮中贮存;采用TRIzol法提取各样品的总RNA,并采用Nanodrop、Agilent 2100方法对RNA样品的纯度、浓度和完整性进行检测,将符合测序要求的槐猪和杜洛克猪的总RNA分别等量混合,形成2个总RNA池,送交北京百迈客生物科技有限公司进行转录组测序(RNAseq)。
1.2 数据整理和分析
1.2.1 原始数据去杂。
测序所获得的原始序列数据均带有一段3′接头序列,并且含有少量低质量序列以及各种杂质成分,经过数据处理得到可用的干净读段(clean reads)。 具体处理方法如下:去除3′接头序列、去除空载读长、去除低质量读段(含有未知碱基N的读段)、去除长度过小或过大的读段等[1]。
1.2.2 序列比对和基因表达量分析。
使用TopHat软件将获得的干净读段(clean reads)与猪参考基因组(Sus scrofa 10.2:ftp://ftp.ensembl.org/pub/release75/fasta/sus_scrofa/)进行比对,并对比对效率进行统计[2]。采用FPKM(fragments per kilobase of transcript per million fragments mapped)作为衡量转录本或基因表达水平的指标[3],FPKM计算公式如下:
FPKM=基因区的读段数目×109基因长度×测序深度(1)
1.2.3 可变剪切预测。
使用Cufflinks软件对基因原有的剪接模型进行比较,进行可变剪接事件(alternative splicing events)预测[3]。常见的可变剪接可分为6种类型:外显子跳跃(exon skipping,ES)、内含子保留(intron retention,IR)、第一个外显子可变剪接(alternative first exon,AFE)、最后一个外显子可变剪接(alternative last exon,ALE)、5′端可变剪接(alternative 5′ splice site,A5SS)、3′端可变剪接(alternative 3′ splice site,A3SS)[4-5]。
1.2.4 基因功能注释及差异表达基因筛选。
使用BLASTX软件将基因序列与Nr、SwissProt、GO、COG和KEGG数据库进行比对,获得所有基因的注释信息。使用R软件的EBSeq包进行基因差异表达分析[6],以=|log2(FC)|≥1且FDR<0.01作为筛选差异基因的标准。其中,差异倍数(fold change,FC)表示两样品之间表达量的比值,错误发现率(false discovery rate,FDR)是对差异显著性P值进行BenjaminiHochberg方法校正后得到的[7]。
1.2.5 GO功能分类。
使用Blast2GO软件对差异表达基因进行GO功能分类,参数e值≤10-5[8],并使用WEGO软件进行统计并绘图。GO(gene ontology)数据库能对基因、蛋白质进行描述,其包含3个本体:细胞组分(cellular component)、分子功能(molecular function)和生物学过程(biological process)。
1.2.6 KEGG通路显著性富集分析。
以KEGG通路为单位,找出与整个基因组背景相比在差异表达基因中显著性富集的通路,其计算公式如下:
P=1-∑m-1i=0
Mi
N-Mn-i
Nn
(2)
式中,N为所有基因中具有通路注釋的基因数目;n为具有通路注释基因中差异表达基因的数目;M为所有基因中注释为某特定通路的基因数目;m为注释为某特定通路的差异表达基因数目。Q≤0.05的通路定义为在差异表达基因中显著富集的通路(Q表示错误比例,Q=V/R,其中R代表所挑选的差异基因个数,V代表没有差异表达的基因个数,即假阳性结果)[1]。
2 结果与分析
2.1 测序数据质量评估
对槐猪和杜洛克猪背部皮下脂肪组织进行转录组测序,获得原始读段数目分别为46 891 554和45 309 436条,原始读段经过去杂处理后,获得的干净读段数分别为37 858 968和37 545 394 条,其中78.54%和82.08% 的干净读段(clean reads)比对上猪参考基因组,可以满足后续分析的需要。
2.2 可变剪切预测
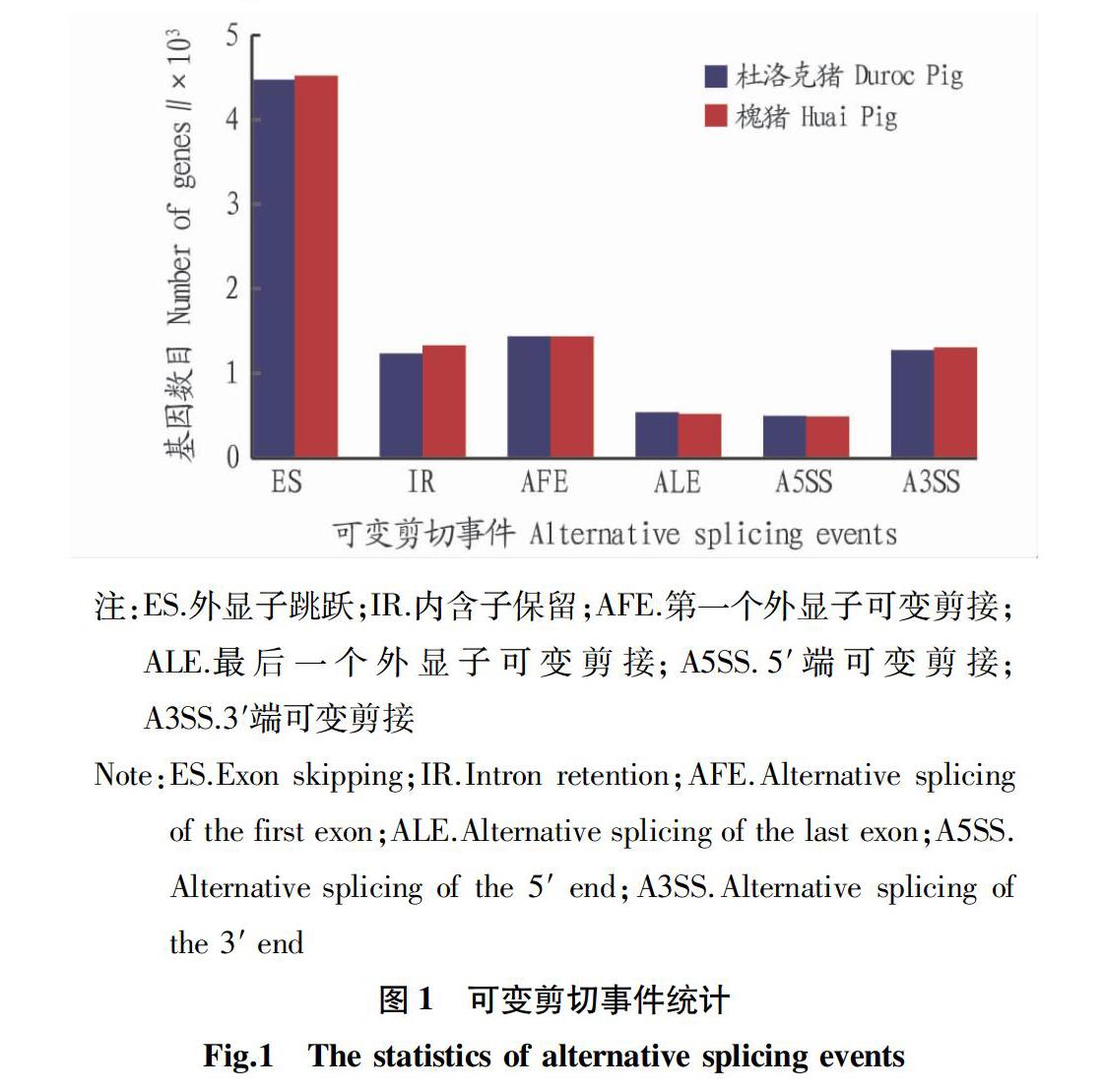
槐猪和杜洛克猪背部皮下脂肪组织分别预测到9 612和9 457可变剪接事件。由图1可知,在6个常见的可变剪切类型中外显子跳跃的数量最多。
2.3 差异表达基因筛选和注释
对槐猪和杜洛克猪背部皮下脂肪组织进行转录组测序,并进行差异表达基因筛选。结果发现,两猪种共有709个差异表达基因,与杜洛克猪相比槐猪有395个上调基因和314个下调基因。各个数据库注释到的差异表达基因数目及所占比例如表1所示。
2.4 GO功能分类 由图2可知,槐猪和杜洛克猪背部皮下脂肪组织的差异表达基因共归类到46个GO条目中,其中细胞组分11个、分子功能12个及生物学过程23个。细胞组分中分类到较多基因的条目为cell part、cell和organelle,分别含540、540和394个基因;分子功能中的条目binding含450个基因;生物学过程中的条目cellular process、metabolic process和biological regulation分别含519、462和412个基因。
2.5 KEGG通路显著性富集分析
槐猪和杜洛克猪背部皮
下脂肪组织之间的差异表达基因共显著富集到9个KEGG通路,通路信息如表2所示。
3 讨论
该研究对槐猪和杜洛克猪背部皮下脂肪组织进行转录组测序,分别获得了37 858 968和37 545 394条干净读段。陈伟[9]对莱芜猪和大白猪背最长肌的转录组测序分别获得40 498 476 和59 072 892条干净读段。Sodhi等[10]对济州岛本地猪和巴克夏猪的脂肪组织进行了测序,分别获得41 411 459 和40 308 452条干净读段。由此可见,对不同猪品种同一个组织测序所获得的干净读段数目不同;对同一个猪品种的不同组织的测序所获得的干净读段数目也不同,这可能是由于不同组织所行使的生理功能和所含的基因数目不同。
该研究在槐猪和杜洛克猪2个品种猪背部皮下脂肪中分别预测到9 612和9 457个可变剪切事件,其中外显子跳跃的数量最多。Kang等[11]在研究不同年龄滩羊皮肤组织的可变剪切时,在1月龄皮肤组织发现有4 120个可变剪切,而在48月龄的皮肤组织中有4 237个可变剪切事件。李青芝等[12]在长白母猪背部皮下脂肪、大网膜脂肪和肠系膜脂肪的转录组研究中发现936~1 029个可变剪切事件。对基因组中可变剪切事件的预测,将有助于理解这些基因在转录调控机制方面的功能。
该研究对槐猪和杜洛克猪背部皮下脂肪组织之间的差异表达基因进行GO功能分类和KEGG通路分析,有助于进一步了解差异表达基因的功能及基因产物在细胞代谢过程中的作用。KEGG通路反映蛋白质之间的互相作用关系,参与代谢通路的蛋白的表达量或者酶的活性改变都可以引起通路调节出现差异。该研究显著富集的通路中,发现一些与调节脂类代谢显著相关通路,如胰岛素信号通路和PPAR信号通路等,为进一步研究脂肪沉积的分子机制提供了一定的参考,后期将结合蛋白质功能试验(如基因敲除、转基因和RNA干扰等)进一步证实。
参考文献
[1] 耿立英,巩元芳,刘铮铸,等.北京油鸡和来航鸡脾脏差异表达基因的筛选与分析[J].中国兽医学报,2017,37(5):941-948,964.
[2] KIM D,PERTEA G,TRAPNELL C,et al.TopHat2:Accurate alignment of transcriptomes in the presence of insertions,deletions and gene fusions[J].Genome Biol,2013,14(4):1-13.
[3] TRAPNELL C,WILLIAMS B A,PERTEA G,et al.Transcript assembly and quantification by RNASeq reveals unannotated transcripts and isoform switching during cell differentiation[J].Nat Biotechnol,2010,28(5):511-515.
[4] 费冰虹.纹枯病菌对寄主侵染的选择性机制初步研究[D].雅安:四川农业大学,2015.
[5] 刘文鑫.杨树溃疡病发生的水分生理效应及转录组分析[D].保定:河北农业大学,2015.
[6] LENG N,DAWSON J A,THOMSON J A,et al.EBSeq:An empirical Bayes hierarchical model for inference in RNAseq experiments[J].Bioinformatics,2013,29(8):1035-1043.
[7] 李国辉.金茅黑鸡A系胸肌转录组特征及对重要肉用性状的影响[D].扬州:扬州大学,2018.
[8] GTZ S,GARCAGMEZ J M,TEROL J,et al.Highthroughput functional annotation and data mining with the Blast2GO suite[J].Nucleic Acids Res,2008,36(10):3420-3435.
[9] 陈伟.莱芜猪和大白猪背最长肌miRNA与mRNA转录组测序及特征分析[D].泰安:山东农业大学,2014.
[10] SODHI S S,PARK W C,GHOSH M,et al.Comparative transcriptomic analysis to identify differentially expressed genes in fat tissue of adult Berkshire and Jeju Native Pig using RNAseq[J].Mol Biol Rep,2014,41(9):6305-6315.
[11] KANG X L,LIU G,LIU Y F,et al.Transcriptome profile at different physiological stages reveals potential mode for curly fleece in Chinese tan sheep[J].PLoS One,2013,8(8):1-9.
[12] 李青芝,王滔,李明洲,等.豬皮下与内脏脂肪组织mRNA转录组的构建与差异分析[J].四川农业大学学报,2013,31(1):65-70.
