
顶空固相微萃取-气相色谱-质谱联用法同时测定湖库水中12种氯苯甲醚的条件优化
2019-12-31 06:08熊茂富任敏杜伊赵高峰王晓燕
岩矿测试 2019年6期
熊茂富, 任敏, 杜伊, 赵高峰, 王晓燕*
(1.首都师范大学资源环境与旅游学院, 北京 100048;2.水利部信息中心, 北京 100053)
氯苯甲醚类化合物(CAs)是茴香醚苯环上的氢原子被氯取代而形成的一氯至五氯代苯甲醚。该类物质具有较低的挥发性和较高的沸点,水溶性差、亲脂性强等物理性质[1-3]。CAs是水中土霉味的主要来源之一[4],其中主要以4-氯苯甲醚(4-CA)、2-氯苯甲醚(2-CA)、2,4-二氯苯甲醚(2,4-DCA)、2,6-二氯苯甲醚(2,6-DCA)、2,3,6-三氯苯甲醚(2,3,6-TCA)及2,4,6-三氯苯甲醚(2,4,6-TCA)对水中土霉味贡献较大。如Nystrom等[5]在检测瑞典的地表水时发现2,4,6-TCA普遍存在,湖水中的2,4,6-TCA是湖水土霉味的主要来源;毛敏敏等[6]对我国某湖泊的研究中发现2,4,6-TCA的浓度值为4.35ng/L,说明这类物质虽然广泛存在,但其浓度值很低,属于痕量物质。CAs类物质在水中具有很低的嗅阈浓度[7],一般在1~50ng/L范围,尤其是TCA的土霉味最为严重,其嗅阈值小于4ng/L,很难对其进行检测。TCA也是葡萄酒中的主要污染物,人类对葡萄酒中TCA的排斥值小于3.1ng/L[8]。然而该类物质并未在工业上大量生产,在地表水中其主要是有氧条件下微生物降解五氯酚(PCP)及其钠盐(Na-PCP)的产物;饮用水中来自氯的消毒过程中,与水中的茴香醚反应,取代苯环上的氢原子,生成CAs。CAs在沿食物链传播过程中,容易在生物体中富集,特别是对于人类这一食物链顶端物种,它的危害性更易被放大,其中五氯苯甲醚(PCA)的毒性最大,近几年PCA已被列入持久性有机污染物之一[9]。
CAs在水中的含量极低(ng/L水平),但该类物质带来的危害大,在国际上已备受关注[10-11]。而关于CAs在水中的限值目前国际上并未给出,对于其前体物挥发性酚类在我国生活饮用水标准中规定不能超过0.002mg/L。对于性质相似的一氯苯,世界卫生组织(WHO)和我国饮用水质量标准限值均为0.3mg/L。目前,国际标准化组织(ISO)也仅给出了软木塞中2,4,6-TCA的检测标准[12],水中CAs的检测也大多以单个物质为目标或代表物,而对于水体中CAs类污染物的方法研究较少,也未给出一致的标准方法。
实验室在检测水中痕量物质时,一般先把目标物质富集之后再进行检测[13]。对于前处理富集技术,国内外学者为了提高富集倍数,提出了很多方法,如吹扫捕集、固相萃取(SPE)、固相微萃取(SPME)。吹扫捕集技术是Bellar等[14]提出的测定水中挥发性物质的富集方法,其原理是使用气流将待测物质吹脱后进入气相色谱仪进行定量分析,然而这种方法的浓缩限值为20~200ng/L,高于一些嗅味物质的嗅阈值,因此,对于嗅阈值较低的物质如CAs不太合适。SPE技术是利用一种特殊的填料装置,在真空机的抽取作用下,样品被吸附到填料上,随后在有机溶剂的作用下样品被重新溶解[15],进而达到富集的目的。其缺点是整个萃取过程所需时间较长,同时所用的萃取填料柱为一次性使用,成本较高。SPME技术是加拿大Arthur等[16]于1990年研发在固相萃取的基础上演变而来的,萃取时只需将萃取纤维暴露在样品中,检测样品被吸附到纤维上,再进行样品的解离后进入相应的仪器中进行检测分析。该技术操作比较简单,耗时少,检出限低且准确度高,萃取过程中无需使用有机溶剂,同时避免了SPE容易堵塞和产生沟流等缺点。王丹华等[17]利用新型固相微萃取涂层准确检测了水中8种多环芳烃。顶空固相微萃取(HS-SPME)实现了将采集、进样萃取及分析过程一体化,无需溶剂,大幅提高了检测效率。Kristiana等[18]在检测饮用水中含氮消毒副产物时使用了HS-SPME方法,检出限在0.9~80ng/L;余胜兵等[19]使用HS-SPME法对饮用水中单个TCA进行富集,取得了较好的检出效果。
水体中CAs一般是以多种痕量同时存在,建立水中CAs的检测方法是分析水中异味物质来源的基础。对于CAs的检测方法,已有气相色谱-质谱法(GC-MS)[20-23]、气相色谱-原子发射光谱联用技术(GC-AES)[24]和生物传感器法[25],其中GC-MS的定性能力强,在浓度较低时仍有较好的响应值[26],同时该方法也具有较好的选择性[27-29]。但已有的研究大多是以单个CAs为目标物,没有给出同时对多种CAs类物质的最佳检测条件。本文在前人研究的基础上,考察了HS-SPME对多种CAs的富集效果,首先采用HS-SPME对水中目标物质进行萃取,再用GC-MS对萃取物质进行检测,建立了湖库水中12种CAs同时测定的检测方法。研究结果可为后续CAs及水体中痕量物质的检测提供技术条件参考。
1 实验部分
1.1 仪器与设备
气相色谱-质谱仪(6890GC/5975MS,Agilent公司,美国);色谱柱DB-5MS(30m×0.25mm×0.25μm,Agilent公司,美国)。萃取头包括:CAR/PDMS(85μm羧乙基/聚二甲基硅氧烷,Supelco公司,美国);50/30μm DVB/CAR/PDMS(1cm,57328-U,Supelco,美国);PA(85μm,57305,Agilent公司,美国)。PC-420D数字型磁力加热搅拌装置(Corning公司,美国);15mL固相微萃取专用样品瓶(Supelco公司,美国)。
电子天平(称重范围220g;精度0.01mg,Sartorius公司,德国);马弗炉(余姚市亚星仪器仪表有限公司)。
1.2 标准样品和溶液配制
12种CAs标准样品(纯度≥98%,Dr.Ehrenstorfer公司,德国);甲醇为农残级(J.T.Baker公司,美国);氯化钠为分析纯(国药集团化学试剂有限公司),使用前置于400℃的马弗炉中烘干2h,干燥器中密闭保存、备用;超纯水(经Milli-Q系统纯化,电阻率为18.2MΩ·cm)。
分别称取1mg(精确到0.01mg)的3-氯苯甲醚(3-CA)、4-CA、 2-CA、2,6-CA、3,5-氯苯甲醚(3,5-CA)、2,4-CA、2,3-氯苯甲醚(2,3-CA)、2,4,6-TCA、2,3,6-TCA、2,3,4-三氯苯甲醚(2,3,4-TCA)、五氯苯甲醚(PCA)固体标准物质,以甲醇溶解定容至100mL,得到上述11种物质的标准溶液(10mg/L)。2,3,5,6-四氯苯甲醚(2,3,5,6-TeCA)为液体标样(10mg/L)。
准确量取上述11种CAs的混合标准溶液和2,3,5,6-TeCA的标准溶液各1mL于100mL容量瓶中,用甲醇定容至100mL,配制成100μg/L的12种CAs的混合标准溶液,摇匀置于4℃冰箱,避光保存。
1.3 样品前处理
取10mL标准溶液于15mL萃取瓶(Supelco)中,加入3.5g 氯化钠和磁性搅拌子(PTFE包覆,2mm×10mm),拧紧瓶盖(衬有PTFE瓶垫),对磁力搅拌器(转速为1150r/min)进行升温,待水浴温度达到恒定90℃时,将萃取瓶置于水浴中,使瓶身2/3的深度没入水中,并将附有85μm CAR/PDMS萃取纤维的手动萃取针插入瓶内(距水面约0.5mm),萃取40min,立即转移至GC进样口,于300℃下解吸附2min。由保留时间、特征离子定性,外标法定量。
1.4 气相色谱-质谱条件
GC条件:载气为高纯氦气,纯度不小于99.999%,恒流模式,流速1.5mL/min;进样方式为不分流进样;升温程序初始温度50℃,保持1min,以5℃/min升温至180℃,保持1min,以20℃/min升温至285℃;解吸时间2min;进样口温度300℃。
MS条件:采用选择性离子扫描模式(SIM)下无分流进样方式进行检测,电子轰击源(EI);电子能量为70eV;离子源温度为230℃;传输杆温度为250℃;溶剂延迟9min;扫描时间为9.0~33.25min。
1.5 质量控制
实验玻璃器皿依次用洗漆剂、重铬酸钾洗液浸泡、自来水、去离子水漂洗,再用烘箱烘干,使用前分别用甲醇、丙酮、二氯甲烷润洗。其中每隔10个样品添加一个溶剂空白和程序空白,避免背景污染,在探索每种因素对萃取效率的影响时,每组实验重复三次以降低实验误差。在优化后的仪器条件下,各物质色谱峰可以良好分离。12种物质在1~50ng/L范围内线性均良好,能够用于定量,以3倍信噪比(S/N)时的浓度为检出限,其中CAs在水中的检出限为0.045~0.185ng/L,相关系数≥0.9978。使用基质加标法测定样品中的回收率,样品回收率为95.5%~115.1%,相对标准偏差(RSD)≤13.02%。
2 结果与讨论
对于HS-SPME来说,在萃取头涂层一定的条件下,目标化合物的吸附量主要取决于两相的分配系数、目标物的传质速率和顶空体积等因素[30]。本文分别对转子转速、萃取温度、萃取时间、离子浓度(氯化钠的用量)等条件进行优化。
2.1 萃取条件优化
2.1.1SPME萃取头的选择
不同的分析物具有不同的挥发性、极性和分子量,在萃取时要尽可能多的吸附不同的目标物质,同时目标物质又能在热解析时快速脱离萃取涂层[31]。本实验对不同的萃取头物化性质及应用范围进行比较,主要考察了三种常用萃取头:CAR/PDMS、DVB/CAR/PDMS、PA的萃取效果。在温度为80℃、0.35g/mL离子浓度、顶空固相微萃取40min条件下,对混合标准溶液进行萃取分析,结果见图1。从实验数据看出,三种萃取头响应值大小并没有很明显的分布规律,CAR/PDMS萃取头对一氯和二氯苯甲醚响应均值大于5000,DVB/CAR/PDMS萃取头对应响应均值大于4000,两种萃取头对三、四及五氯苯甲醚萃取响应值明显降低,PA萃取头则更低。这与目标物质随分子质量增加挥发性降低有关。CAR/PDMS主要用于挥发性物质、胺类、硝基芳香类化合物的萃取;DVB/CAR/PDMS主要用于具有香味的挥发性化合物;PA萃取头主要用于极性较强的挥发性物质。CAR/PDMS采用多孔渗水,是一种聚合体材料,对极性和非极性化合物有较强的吸附能力。在以往研究中,魏魏等[32]在对水中包括CAs在内的多种嗅味物质进行测定时选用了CAR/PDMS作为萃取头,对目标物质的吸附能力强,萃取效率高,故后续分析实验均使用CAR/PDMS萃取头。
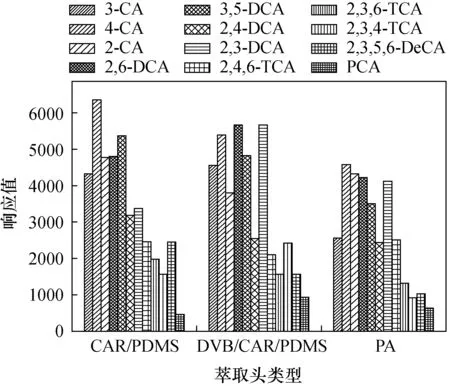
图1 不同萃取头对萃取效果的影响Fig.1 Effect of different fiber typers on the extraction efficiency
2.1.2萃取温度优化
体系温度影响到整个萃取过程中的热力学和动力学。在一定温度下溶液体系达到气液平衡,气相组成与样品原来组成成正比关系。随体系温度升高,气相中目标分析物浓度变大,有利于纤维的吸附。但温度过高会降低分配系数,从而降低回收率[33],因此需要对萃取温度进行优化。

图2 萃取温度(a)、离子浓度(b)、萃取时间(c)和搅拌速度(d)对萃取效率的影响Fig.2 Effect of extraction temperature(a), ion concentration(b), extraction time(c) and mixing speed(d) on extraction efficiency
取标准液10mL于15mL顶空瓶中,加入3.5g氯化钠,搅拌速率为1150r/min,萃取时间40min条件下。考察了萃取温度(20℃、40℃、60℃、80℃、90℃)对12种CAs萃取效率的影响(图2a)。当温度从20~60℃时,其响应值变化不大;当温度到达80℃时响应值大幅增加,在80℃之后其变化不明显,可能因为达到一定温度后,CAs发生了解析现象。从图中的变化趋势可以看出温度大于80℃后,大部分目标物质响应值都有所上升,而 4-CA、2-CA、2,6-DCA及2,4,6-TCA的响应值有少许下降,可能原因是温度过高导致热解析,使目标物质从萃取纤维上脱落下来,进而降低萃取效率。参考以往研究,余胜兵等[19]对水中嗅味物质研究中,选择萃取温度为70℃;徐振秋等[34]在测定饮用水中嗅味物质时萃取温度为60℃。对于CAs可能随着CAs物质相对分子质量的增加,对萃取温度的要求明显提升。因此,为了充分萃取目标物质,选择萃取温度为80℃。
2.1.3离子强度优化
样品中加入盐后,盐在水中电离出的离子占据了有机物周围的水分子,降低了目标物质的溶解度,使得目标物分子较多地挥发至顶部空间,从而有利于CAs在萃取头上的吸附,提高了方法灵敏性[35]。本研究中选择NaCl增加离子浓度。在15mL的顶空瓶中加入10mL标准溶液,萃取温度为80℃,萃取时间选择40min,搅拌速率为1150r/min。考察NaCl加入量(0、0.10、0.20、0.35、0.40g/mL)对12种CAs萃取效率的影响(图2b)。结果表明,随着NaCl加入量的增加,大部分目标物的峰面积先减小再增大后变化不明显。开始峰面积减小是由于加入少量NaCl对CAs的竞争吸附;然后峰面积增大是由于盐析效应;最后峰面积变化不明显是由于NaCl在水中溶解趋于饱和,盐析效应减小,此外过高的离子强度对CAs分子竞争吸附的同时,也会破坏CAs的理化性质,从而降低CAs的传质速率[36],影响萃取效率。离子浓度为0.35g/mL时,达到饱和,各目标物的峰面积最大,这与宋荣娜等[37]采用相同的方法测定水中异味物质时得出的最优离子浓度一致,所以本实验选择离子浓度为0.35g/mL。
2.1.4萃取时间优化
萃取时间是判定待测物在基质和固定相之间达到平衡的重要因素,选择合适的时间不仅能提高萃取效率,还能减少样品由于长时间处于萃取环境中而带来的纯度的降低,时间太短,萃取不完全,时间过长,会延长分析时间,降低工作效率,萃取时间过长,还可能发生目标物的脱附[38]。本研究考察10、20、30、40、50min不同萃取时间对12种CAs萃取效率的影响(图2c)。结果表明:10~40min,随萃取时间增加,峰面积增加;40~50min,峰面积略有下降,相似的趋势在以往相关研究中也有被发现[39],萃取过程中基质和固定相之间需达到同一动态平衡状态,同一平衡状态不会对SPME的定量造成影响,同时考虑到对目标物质的充分萃取,故选定萃取时间为40min。
2.1.5搅拌速率优化
对样品的搅拌有助于目标物和涂层间的传质,使样品均匀,增加萃取针和目标物的接触,提高萃取效率,同时还可以促进NaCl的溶解,使平衡时间缩短。本研究考察了搅拌速度0、200、400、600、800、1150r/min对12种CAs萃取效率的影响(图2d)。结果表明,随搅拌速率增加,响应值亦增加,当达到仪器最大搅拌速度时,萃取效率也最高,本次实验没有对后续搅拌速度进行对比分析,这一趋势与马康等[40]的研究结果一致,同时考虑到萃取纤维易变形问题,因此选定1150r/min为实验最优搅拌速度。
2.1.6样品体积优化
在萃取温度不变的情况下,体系的平衡常数维持不变[41]。样品量一定时气相与液相的相比相同,因此样品体积是影响分析结果的重要因素之一。减小样品体积,可以增大顶空体积,使CAs在两相间的分配平衡偏向顶空气相,使更多的CAs挥发至顶空;然而顶空体积过大,也会造成CAs浓度的稀释,使萃取效率降低[42]。普遍认为,在萃取温度不变的情况下,随着顶空体积变小,萃取效率一般会变高。本研究考察了在15mL样品瓶中加入4、6、8、10mL标准液对12种CAs萃取效率的影响(图3)。结果表明,样品体积在4~6mL时,随样品量的增加,峰面积迅速增大;在样品体积为6~10mL时,峰面积也在增加,但增加的幅度较小。当样品量高于10mL时,可能会导致萃取纤维接触水相,破坏萃取纤维,故选择样品量为10mL。

图3 样品体积对萃取效率的影响Fig.3 Effect of sample volume on extraction efficiency
2.2 方法技术指标
2.2.1检出限
本实验采用顶空固相微萃取技术,其中确定的最优萃取条件为萃取时间40min,离子浓度0.35g/mL,萃取温度80℃,样品用量10mL(15mL瓶),解析时间2min。将100μg/L的混合标准母液稀释为1、5、10、20、50ng/L的5种浓度12种物质的混合标准水样,在优化后的仪器条件下,各物质色谱峰可以良好分离。12种物质在1~50ng/L范围内线性良好,相关系数在0.9978以上。当置信水平为99%时,以3倍信噪比(S/N)时的浓度为检出限(表1)。毛敏敏等[6]采用固相微萃取法对水样中TCA进行检测的检出限为0.24ng/L,本实验采用顶空固相微萃取TCA的检出限均小于0.185ng/L。
2.2.2准确度和精密度
准确度是指测定结果与真实值之间接近的程度,常用回收率评价方法的准确度,用相对标准偏差评价方法的精密度[43]。依据样品加标回收率原理,用10、50ng/L两种组分混合标准溶液进行加标实验。按上述优化的萃取方法和仪器条件进行回收率实验,每个水平重复6次,结果见表2。表中对12种目标分析物进行回收率及相对标准偏差(RSD)分析,目标物平均回收率在95.5%~115.1%之间,具有较好的准确度;RSD均值在5%以下,说明实验的操作及仪器的精密度具有良好的可靠性。沈斐等[44]采用吹扫捕集结合GC-MS法测定饮用水中嗅味物质时,加标回收率在71.0%~125.0%之间。唐熙等[45]使用甲醇超声萃取结合GC-MS测定软木塞中土霉味物质的加标回收率在88.4%~97.6%之间。因此,本实验回收率(95.5%~115.1%)表明该方法对于CAs的适用度更佳。
表1氯苯甲醚的线性关系和方法检出限
Table 1 Linear relationship and detection limits of CAs
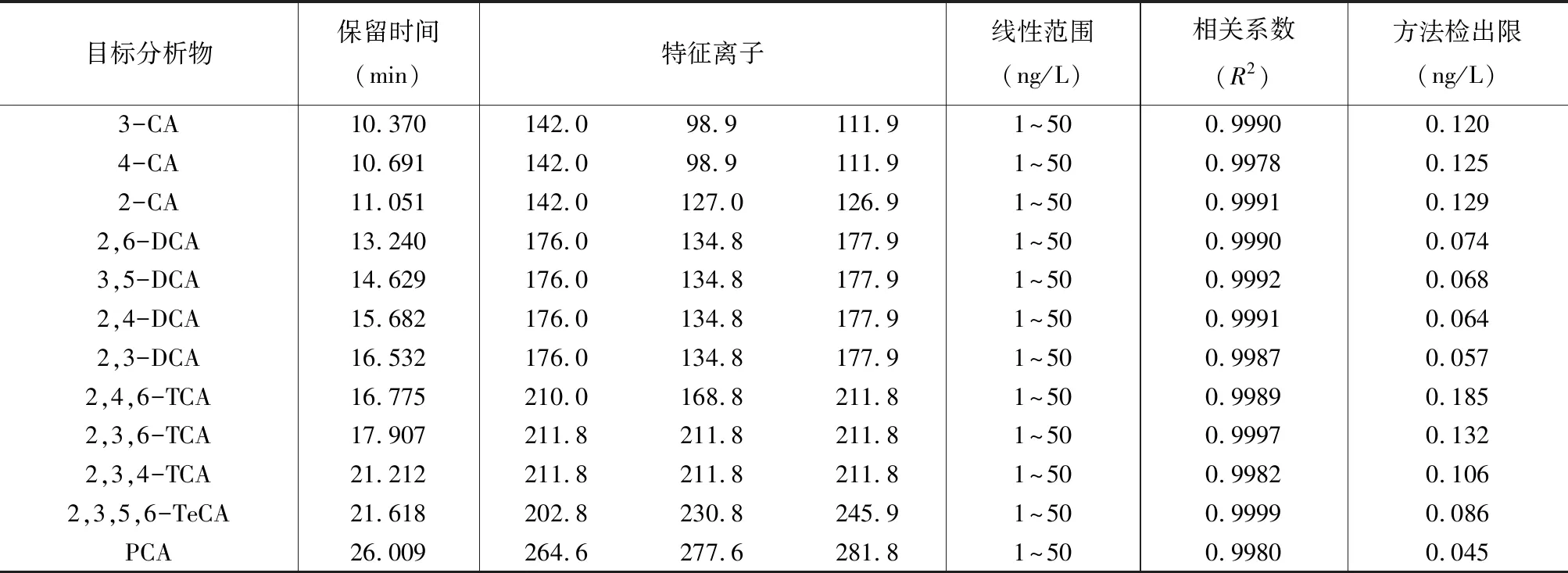
目标分析物保留时间(min)特征离子线性范围(ng/L)相关系数(R2)方法检出限(ng/L)3-CA10.370142.098.9111.91~500.99900.1204-CA10.691142.098.9111.91~500.99780.1252-CA11.051142.0127.0126.91~500.99910.1292,6-DCA13.240176.0134.8177.91~500.99900.0743,5-DCA14.629176.0134.8177.91~500.99920.0682,4-DCA15.682176.0134.8177.91~500.99910.0642,3-DCA16.532176.0134.8177.91~500.99870.0572,4,6-TCA16.775210.0168.8211.81~500.99890.1852,3,6-TCA17.907211.8211.8211.81~500.99970.1322,3,4-TCA21.212211.8211.8211.81~500.99820.1062,3,5,6-TeCA21.618202.8230.8245.91~500.99990.086PCA26.009264.6277.6281.81~500.99800.045
表2氯苯甲醚的回收率及相对标准偏差
Table 2 Recoveries and relative standard deviations of CAs
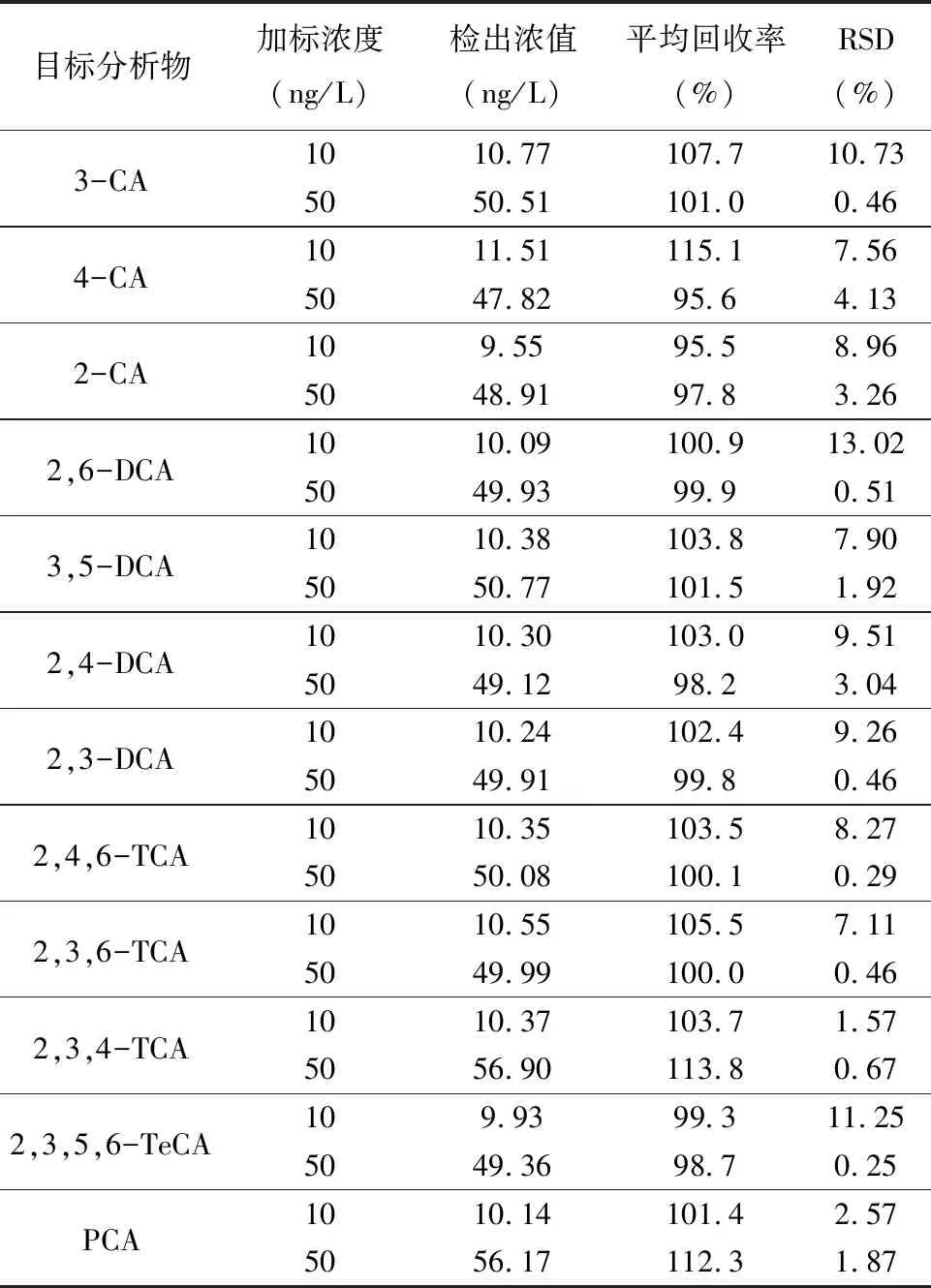
目标分析物 加标浓度(ng/L)检出浓值(ng/L)平均回收率(%)RSD(%)3-CA1010.77107.710.735050.51101.00.464-CA1011.51115.17.565047.8295.64.132-CA109.5595.58.965048.9197.83.262,6-DCA1010.09100.913.025049.9399.90.513,5-DCA1010.38103.87.905050.77101.51.922,4-DCA1010.30103.09.515049.1298.23.042,3-DCA1010.24102.49.265049.9199.80.462,4,6-TCA1010.35103.58.275050.08100.10.292,3,6-TCA1010.55105.57.115049.99100.00.462,3,4-TCA1010.37103.71.575056.90113.80.672,3,5,6-TeCA109.9399.311.255049.3698.70.25PCA1010.14101.42.575056.17112.31.87
3 结论
本文针对湖库水体中12种CAs的HS-SPME萃取影响因素进行了优化,给出了最佳实验条件。采用HS-SPME结合GC-MS法,实现了对水中12种CAs物质的同时、精确检测,提高了检测效率。该方法的检出限、回收率及相对标准偏差均能满足水体样品的检测要求,可以广泛地应用到水体样品中CAs的检测。因实验中未考虑其他几种CAs,后续研究有必要探究其他几种物质对整个萃取检测过程的影响。
以往研究只针对单个CAs为目标物,而实际水体中CAs是以多物质混合存在的,本研究能为后续工作的开展以及水中痕量物质的检测提供很好的技术参考和实际应用潜力。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31
波谱学杂志(2022年2期)2022-06-14
环境卫生工程(2021年1期)2021-03-19
天然产物研究与开发(2018年6期)2018-07-09
中华皮肤科杂志(2018年6期)2018-01-19
河南农业科学(2017年8期)2017-09-01
现代检验医学杂志(2015年1期)2015-02-06
中国医药导报(2015年36期)2015-01-19
浙江科技学院学报(2014年6期)2014-02-28
中国氯碱(2014年10期)2014-02-28
