
水热法制备Mg/Sn/W 复合氧化物催化剂及其性能评价
2019-12-31 00:44:54汪义超陈亚春张光旭叶云涛
石油化工 2019年12期
汪义超,陈亚春,张光旭,叶云涛
(武汉理工大学 化学化工与生命科学学院,湖北 武汉 430070)
ε-己内酯是一种应用广泛的有机中间体,合成的聚己内酯可生物降解,在生物、医学和环保等领域具有重要的作用[1-3]。目前,由于均相催化剂体系存在许多限制,如催化剂分离回收、ε-己内酯纯化等[4],故非均相催化剂催化氧化环己酮合成ε-己内酯国成为内外研究的热点。如铝/镁水滑石及水滑石类化合物[5-8]、二氧化硅负载锡[9]、与相关金属复配的沸石分子筛[10-11]、Sn-坡缕石[12]、MgO/SnO2、WO3/AlO3复合氧化物[13-14]、天然矿石[15]等作为催化材料,都具有良好的催化性能。
为尽早实现ε-己内酯的工业化,本课题组采用共沉淀法制备出Mg/Sn/W 复合氧化物催化剂[16],在工艺上采用三段式反应精馏合成ε-己内酯,将间歇反应转变为连续反应,以提高ε-己内酯生产效率,并研制出活性较高的填料型催化剂用于反应精馏装置[17-18]。但由于共沉淀法制备出的Mg/Sn/W 粉末催化剂成型后的填料型催化剂存在活性组分W 易流失的问题,造成ε-己内酯的合成难以实现连续化,故如何有效防止W 的流失成了研究的重点之一。
本工作在制备前体过程中加入了分散剂,利用水热法制备了Mg/Sn/W 催化剂,考察了不同种类的分散剂和分散剂含量对催化剂活性的影响;使用N2吸附-脱附、XRD、FTIR、SEM 等分析手段对新型催化剂的结构进行表征,探究了共沉淀法和水热法制备的催化剂在使用过程中W 的流失情况。
1 实验部分
1.1 试剂
SnCl4·5H2O、H2WO4、MgCl2·6H2O、氨水、2-甲基吡啶、乙酸丁酯、乙酸、环己酮、三乙醇胺(TEOA)、聚乙二醇(PEG,相对分子质量1 500)、十六烷基三甲基溴化铵(CTAB)、无水乙醇:分析纯,国药集团化学试剂有限公司;过氧化氢:50%(w),工业纯,河南中原大化集团有限公司。
1.2 水热法制备Mg/Sn/W 复合金属氧化物催化剂
取14 g MgCl2·6H2O 和3.6 g SnCl4·5H2O 及适量的H2WO4置入三颈烧瓶中,再分别加入适量不同种类的分散剂和200 mL 的去离子水,在45℃下,机械搅拌10 min,得橙黄色溶液,逐滴加氨水至pH=10,得乳白色悬浮液,在45 ℃下继续搅拌1 h,将溶液转移至带聚四氟乙烯内衬的水热釜中,密封,放入马弗炉中于180 ℃下保温6 h,取出反应后的溶液进行抽滤,洗涤6 次(用200 mL 蒸馏水洗涤3 次,200 mL 无水乙醇洗涤3 次),然后在恒温干燥箱中100 ℃下干燥12 h。将干燥后的固体用粉碎机粉碎,于马弗炉中焙烧,得到Mg/Sn/W 复合金属氧化物催化剂。将不加任何分散剂(空白)、加入单一分散剂PEG、加入单一分散剂CTAB、加入混合分散剂PEG 和TEOA(质量比为1∶1)、加入混合分散剂CTAB 和TEOA(质量比为1∶1)制备的催化剂分别记为1#,2#,3#,4#,5#。
1.3 分析方法
采用美国Micromeritics 公司TriStar Ⅱ3020 型N2吸附-脱附仪测试催化剂的比表面积及孔体积。采用日本理学公司D/MAX-RB 型转靶X 射线衍射仪表征催化剂的晶体结构,测试条件:Cu Kα射线,管电压为40 kV,功率3 kW,管电流40 mA,扫描速率 2(°)/min,扫描范围2θ=5°~70°。采用美国Thermo Fisher 公司Nicolet Nexus 型傅里叶变换红外光谱仪测定催化剂结构,扫描范围400 ~4 000 cm-1。采用美国赛默飞世尔科技有限公司ESCALAB 250Xi 型X 射线光电子能谱仪表征催化剂表面元素的价态及含量。采用德国蔡司股份公司JEM-7500F 型场发射扫描电子显微镜表征催化剂形貌,采用荷兰PANalytical B V 公司Zetium 型X 射线荧光光谱仪定量分析催化剂中各元素含量。
1.4 催化剂的活性评价
催化剂的活性评价步骤:1)分别称量96 g 乙酸、75 g 乙酸丁酯和1 mL 的2-甲基吡啶加入到带有分水器、恒压漏斗、温度计冷凝管的三口烧瓶中,加入水热法制备的催化剂1.5 g,保持一定的转速,调节水浴温度至55 ℃,真空度保持100 kPa,再将54.4 g 50%(w)的H2O2加入恒压漏斗中,控制液滴缓慢滴入,保持乙酸丁酯与水的共沸物回流,反应5 h 后,制得含有过氧乙酸的混合反应液,将分出的水由分水器底部排出。2)调节水浴温度至60 ℃、在真空度为60 kPa 左右的条件下,加78.4 g 环己酮进入恒压漏斗,控制液滴缓慢滴加到含有过氧乙酸的混合反应液的三口烧瓶,回流反应一定时间,得到含有产物ε-己内酯的混合溶液。3)将混合溶液称量并取样,采用美国Agilent 公司7890B 型气相色谱仪分析试样,根据式(1)~(3)计算环己酮的转化率(XCYC)、ε-己内酯选择性(SC6H10O2)和ε-己内酯收率(YC6H10O2)。
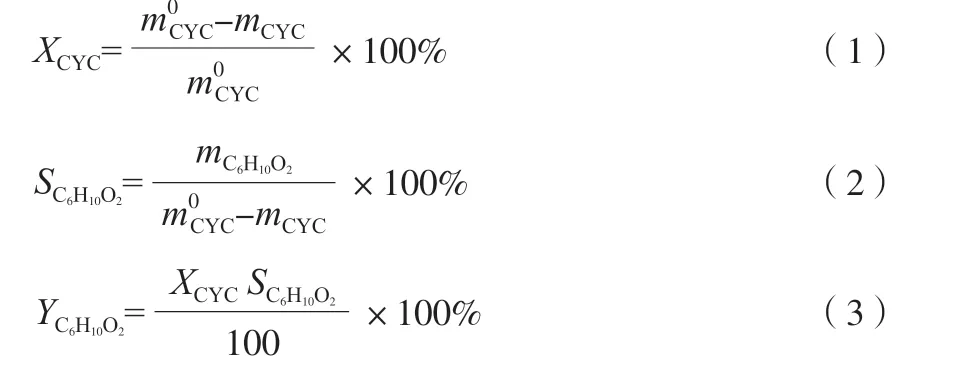
式中,m0CYC为环己酮的初始质量,g;mCYC为环己酮剩余质量,g;mC6H10O2为ε-己内酯的生成质量,g。
2 结果与讨论
2.1 催化剂的表征结果
2.1.1 XRD 表征结果
图1 为水热法合成的Mg/Sn/W 催化剂的XRD谱图。从图1a 可看出,加入分散剂后,催化剂均出现了Mg2SnO4(PDF 24-723)、MgSnO3(PDF 30-798)、MgWO4(PDF 52-390)和WO3(PDF 20-1323)的特征峰,与不加分散剂的催化剂相比,加入分散剂的催化剂衍射峰强度增加,结晶度变好;且采用混合分散剂CTAB 和TEOA 比采用单一分散剂制得的催化剂衍射峰强度更高,结晶度增大;而采用PEG 和TEOA 混合分散剂制得的催化剂比采用单一分散剂制得的催化剂衍射峰强度变弱、结晶度变差,说明该材料存在结晶缺陷,进而导致催化剂活性增加。从图1b 可看出,使用后催化剂的衍射晶型与新鲜催化剂相比并无明显变化,除了在2θ=33.8°,42.6°处的MgSnO3的衍射峰强度稍有下降,其他晶体衍射峰的强度未发生明显变化,说明使用后的催化剂晶体结构并未被破坏,能大致保持原有催化剂的结构特点。
2.1.2 FTIR 表征结果
图2 为水热法合成的Mg/Sn/W 催化剂的FTIR谱图。
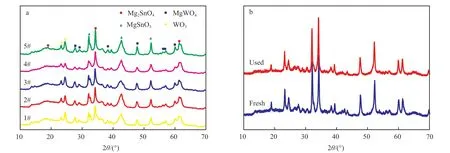
图1 不同分散剂制备催化剂(a)与使用前后4#催化剂(b)的XRD 谱图Fig.1 XRD patterns of catalysts with different dispersants(a) and catalysts of 4# with fresh and used(b).
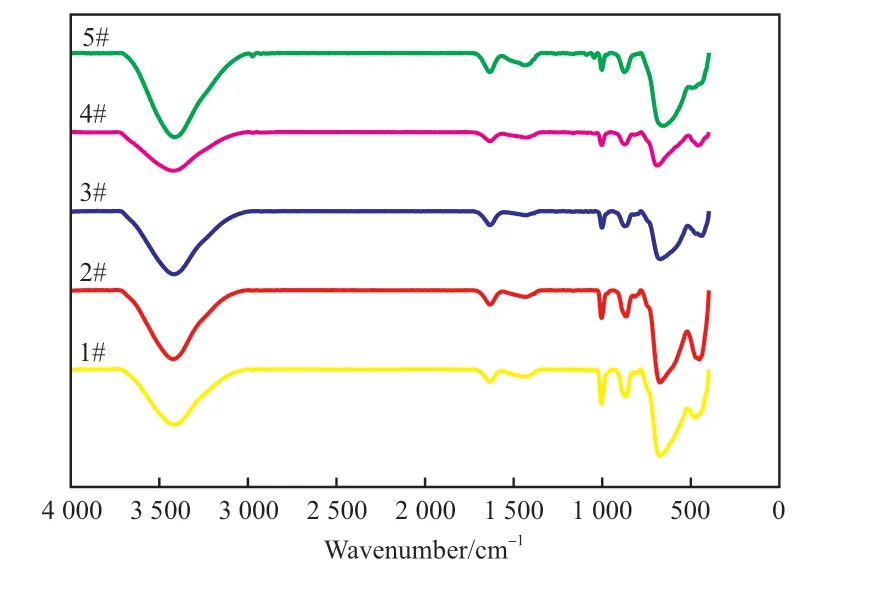
图2 水热法合成Mg/Sn/W 催化剂的FTIR 谱图Fig.2 FTIR spectra of Mg/Sn/W catalysts by hydrothermal methods.
从图2 可看出,不加分散剂与加入分散剂制备的催化剂相比,水热法合成Mg/Sn/W 催化剂的吸收峰相似,在3 425 cm-1处出现水分子或O—H 键的伸缩震动峰,在1 630 cm-1处出现较弱的吸收峰是由于水分子的H—O—H 键之间的弯曲振动,这说明催化剂中可能存在着结合水。在1 000 cm-1和850 cm-1处出现的吸收峰分别由W=O 和W—O—W 键的伸缩振动引起,在676 cm-1处出现的吸收峰和Sn—O 键的振动有关,这些离子键的存在说明催化剂的活性和钨酸盐及锡酸盐成分的组成有很大的关系。
2.1.3 XPS 表征结果
对4#催化剂进行XPS 表征分析,并对催化剂表面的各元素含量及价态进行研究,结果见图3。从图3a 可看出,谱图中两个峰分别对应Sn 3d3/2和Sn 3d5/2,表明Sn 元素在催化剂内主要价态是正四价,说明Sn 元素是以锡酸盐或二氧化锡的形式存在。从图3b 可看出,谱图中的两个峰分别对应于W 4f7/2和W 4f5/2,因此催化剂中的W 元素仅以正六价的方式存在,表明氧化钨在催化剂中为主要的给电子体。以最高价态存在的Sn 和W 元素具有额外的电子,有助于催化剂中电子的传递,提高催化剂的催化活性[17]。从图3c 可看出,谱图中的两个峰对应于化学吸附氧Oα和晶格氧Oβ,二者的比例 分别是51.44%和48.56%,说明吸附氧含量较大。
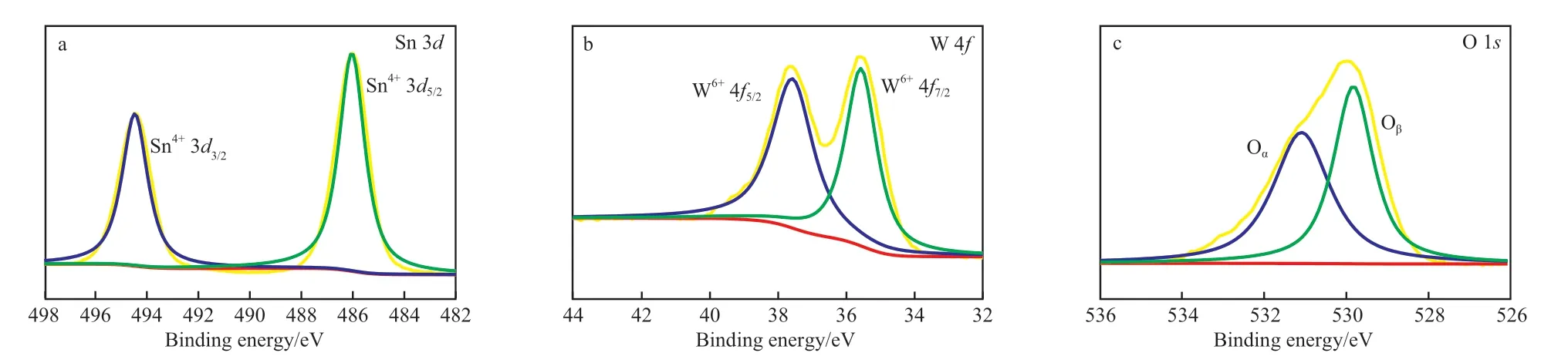
图3 水热法合成Mg/Sn/W 催化剂的XPS 谱图Fig.3 XPS patterns of Mg/Sn/W catalysts by hydrothermal method.
2.1.4 SEM 表征结果
图4 为水热法合成Mg/Sn/W 催化剂的SEM 照片。从图4 可看出,水热法制备的Mg/Sn/W 催化剂呈片状颗粒。没有加分散剂的催化剂(图4a)有较明显的团聚现象;加入PEG 的催化剂(图4b)团聚现象改善,且颗粒变小;加入CTAB 的催化剂(图4c)颗粒变小,但依然存在些许团聚;加入混合分散剂CTAB 和PEG 的催化剂(图4e)的分散效果更好;加入混合分散剂PEG 和TEOA 后合成的催化剂(图4d)的颗粒分散性最好,颗粒更小。

图4 水热法合成Mg/Sn/W 催化剂的SEM 照片Fig.4 SEM images of Mg/Sn/W catalysts by hydrothermal method.
2.2 催化剂的性能
2.2.1 分散剂种类的影响
不同分散剂种类合成催化剂的结构特性见表1。从表1 可看出,加入不同的分散剂对催化剂的形态和结构有较大影响。加入分散剂后催化剂的比表面积明显增大,其中,以PEG 和TEOA 为混合分散剂的催化剂拥有更大的比表面积和更小的孔径,且孔体积最大。
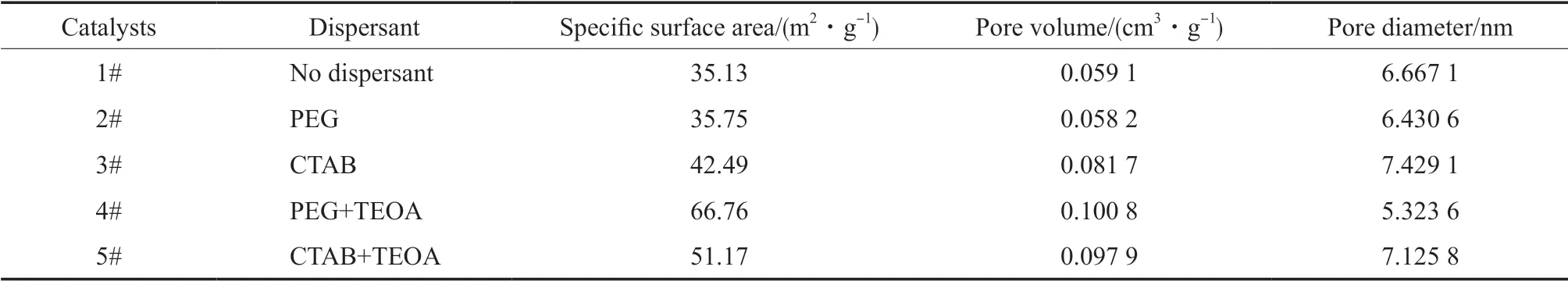
表1 不同分散剂种类合成催化剂的结构特性Table1 Textural properties of catalysts with different dispersants
分散剂种类对催化剂活性的影响见表2。从表2 可知,加入分散剂后制备的催化剂活性提高,表明加入分散剂,改善了催化剂的结构,增大了催化剂的比表面积,从而提高了催化剂生成ε-己内酯的活性,其中,加入混合分散剂PEG 和TEOA 后催化剂的催化活性最大,收率可达56.88%。这可能是因为PEG 作为一种非离子型表面活性剂,在溶液中稳定性高[19],起到很好的自组装、降低催化剂制备过程中的无序沉聚作用[20];而TEOA 可以与溶液中的Mg2+和Sn4+进行络合反应,避免了粒子之间的沉淀,使得悬浮液分散性更好,从而使催化剂的活性提高。
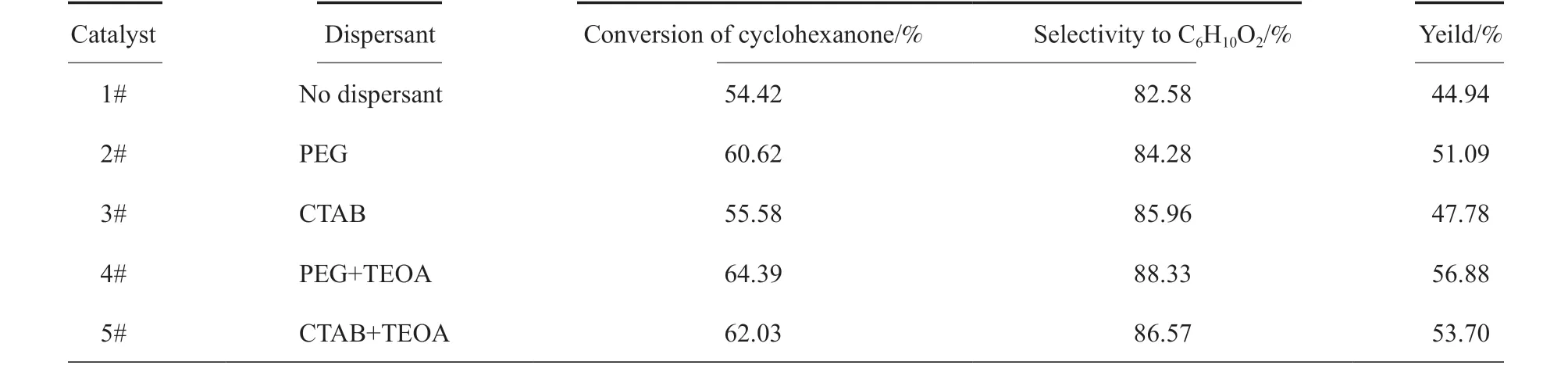
表2 分散剂种类对催化活性的影响Table 2 Effect of dispersants on the activities of catalysts
2.2.2 混合分散剂含量的影响
考察了水热法制备Mg/Sn/W 催化剂过程中混合分散剂含量对催化剂活性的影响,结果见图5。
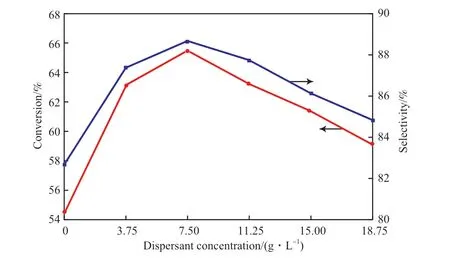
图5 分散剂含量对催化剂活性的影响Fig.5 Influence of dispersant concentration on the activities of catalysts.
从图5 可看出,随着混合分散剂含量的增加,环己酮转化率和ε-己内酯的选择性都是先增加后减少。当混合分散剂的含量为7.5 g/L 时,催化剂的活性最大,收率达58.12%;当加入的混合分散剂的含量大于7.5 g/L 时,收率逐步降低。
为解释此种现象,将不同含量分散剂制备的催化剂进行结构分析,结果见表3。从表3 可看出,适量的混合分散剂可增大催化剂的比表面积,但当含量过大时,催化剂的比表面积会减小,孔体积减小,孔径增大,导致活性位点降低。而活性位点下降的原因可能有两个:1)当分散剂的加入量恰好将颗粒表面包裹时,分散剂就能很好地发挥位阻稳定和静电稳定作用,当超过此用量时,会出现过饱和吸附现象,使悬浮液的稳定性变低[21-23];2)随着分散剂含量增大,孔结构变得规整,比表面积变大,催化活性增加,但当分散剂添加量过多时,在煅烧过程中有机物会产生大量的CO2和H2O,使原本的孔道结构部分坍塌[24],导致催化剂活性下降。
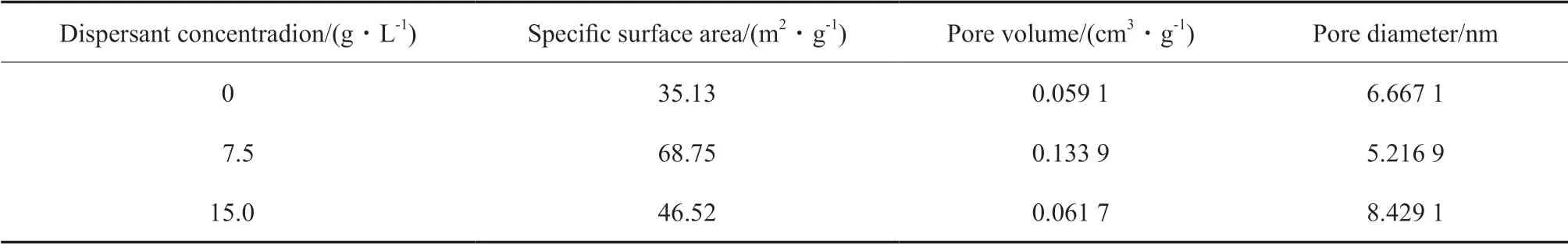
表3 不同含量混合分散剂合成催化剂的结构特性Table3 Textural properties of catalysts with different mixing dispersant concentrations
2.2.3 活性组分W 含量的影响
在混合分散剂含量为7.5 g/L 条件下,活性组分W 含量对催化剂活性的影响见图6。从图6 可看出,随着W 含量的增加,环己酮转化率和ε-己内酯选择性先增加后下降。在钨酸质量浓度为6 g/L 时,催化剂的活性达到最大,环己酮转化率达66.23%,ε-己内酯选择性达89.56%,在钨酸质量浓度超过6 g/L 后,催化剂活性有所下降。这是因为不同的金属离子对过氧乙酸的分解速率不同,Cr3+会促进过氧乙酸的分解[25],作为同一族的W3+具有相似的性能,随着W3+的增加,过量的W3+使过氧乙酸加速分解,产生副产物,导致环己酮转化率和ε-己内酯的选择性下降。这与李莹等[26]的报道相一致,因此选择适量的W 含量在催化反应中有着重要的作用。
2.2.4 共沉淀法和水热法制备的Mg/Sn/W 催化剂的性能比较
将共沉淀法和水热法制备的Mg/Sn/W 催化剂在相同的反应条件下进行活性评价实验。结果见表4。从表4 可看出,由两种方法制备的新鲜催化剂中MgO、SnO2和WO3的含量大致相同;水热法制备的Mg/Sn/W 催化剂合成ε-己内酯的活性更高,从催化剂使用的情况可以发现,水热法制备的催化剂可以有效解决活性组分W 的流失问题,也能在一定程度上保持催化剂活性,使得催化剂的重复利用次数增多。
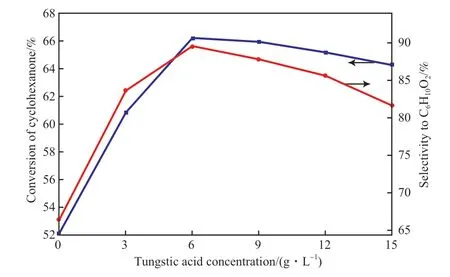
图6 W 含量对催化剂活性的影响Fig.6 Effect of W content on the catalyst activity.
3 结论
1)以PEG 和TEOA 为混合分散剂制备的催化剂拥有更大的比表面积和更小的孔径,改善了催化剂的结构,增大了催化剂的比表面积,提高了催化剂生成ε-己内酯的活性。
2)当混合分散剂的含量为7.5 g/L、在钨酸质量浓度为6 g/L 时,催化剂的活性最大,环己酮转化率达66.23%,ε-己内酯选择性达89.56%。
3)与共沉淀法制备Mg/Sn/W 催化剂相比,水热法制备的Mg/Sn/W 催化剂合成ε-己内酯的活性更高,可以有效解决活性组分W 的流失问题,也能在一定程度上保持催化剂活性。
猜你喜欢
陶瓷学报(2021年5期)2021-11-22 06:35:00
化工管理(2021年7期)2021-05-13 00:45:22
天津造纸(2016年1期)2017-01-15 14:03:29
石油炼制与化工(2016年4期)2016-04-06 20:19:54
天津科技大学学报(2016年1期)2016-02-28 16:59:46
合成化学(2015年10期)2016-01-17 08:55:57
橡胶工业(2015年9期)2015-08-29 06:40:44
四川师范大学学报(自然科学版)(2015年1期)2015-02-28 14:07:30
应用化工(2014年4期)2014-08-16 13:23:09
河南科技(2014年10期)2014-02-27 14:09:08
