
超高压下大豆亲脂蛋白-羟丙基甲基纤维素理化性质研究
2019-12-31 07:52:28钟明明谢凤英孙禹凡齐宝坤
农业机械学报 2019年12期
李 杨 钟明明 廖 一 谢凤英 孙禹凡 齐宝坤
(东北农业大学食品学院, 哈尔滨 150030)
0 引言
大豆亲脂蛋白(Lipophilic protein,LP)的主要成分是油体蛋白和磷脂,它是油体的原始成分,且具有较强的表面活性。LP作为一种营养价值较高的植物蛋白被广泛应用于食品工业中[1]。但在加工中天然大豆蛋白的功能会受到一定程度的破坏,尤其是当溶液的pH值接近蛋白质的等电点时,蛋白质的各种功能都会大幅度下降,因此常对蛋白进行适当的改性处理[2]。研究人员通过多种方式,如加热、超声、微波、超高压等技术手段,改变蛋白质的空间结构和功能特性[3-6],使其表现出更好的功能特性。近年来,天然大豆蛋白改性成为研究热点,如引入磷脂等生物小分子或多糖等生物大分子物质,使其与蛋白质发生相互作用,复合体系的形成对大豆蛋白的功能性质产生重要影响。
羟丙基甲基纤维素(Hydroxypropyl methylce-llulose,HPMC)是一种可食用性纤维素,其健康无毒,且具有良好的水溶性、成膜性和较强的表面活性,可以控制表面压力并改善薄膜粘弹性[7],可作为稳定剂用于乳液制备。作为一种两性离子表面活性剂,HPMC可以通过结合的方式使LP的表面活性发生改变,且它们之间的交互作用会影响LP的功能性质。已有研究通过加入多糖改变蛋白连续相的流变特性和界面膜结构,增强体系的稳定性[8-10]。文献[11]研究表明,多糖连接到蛋白质表面,部分结构可以伸入两相中形成有效的乳液保护层,降低界面张力,增强液滴间静电排斥或空间位阻。但未能清晰地解释超高压处理等物理加工方式对蛋白质-多糖交互作用的影响。
本文通过超高压处理改变蛋白质构象,促进柔性结构展开、刚柔性区域重排、非极性基团暴露,同时改变蛋白质三、四级结构,促进多糖与蛋白质分子的键合,使蛋白-多糖复合物有效提高乳液界面活性、构象稳定性和乳化稳定性;同时,研究超高压处理对LP-HPMC复合程度及复合物功能性质的影响,实验研究超高压处理条件对LP-HPMC复合物形成的影响,以期为超高压技术运用于LP-HPMC复合产品和其他蛋白与HPMC复合的食品加工过程提供理论依据。
1 材料与方法
1.1 材料与试剂
大豆(东农43),东北农业大学大豆研究所;羟丙基甲基纤维素(Ⅰ型,粘度:30 mPa·s),天津市东丽区天大化学试剂厂;葵花籽油,市售;氯化钠,天津市光复精细化工研究所;硫酸,北京新光化工试剂厂;磷酸二氢钠,天津市东丽区天大化学试剂厂;磷酸氢二钠,天津市东丽区天大化学试剂厂;其他试剂均为分析纯。
1.2 仪器与设备
ULTRA-TURRAX UTL2000型高压均质机,上海标本模型有限公司;LGR20-W型台式高速冷冻离心机,北京京立离心机有限公司;Mastersizer2000型激光粒度仪,英国马尔文仪器有限公司;DELTAVISION OMX SR型超分辨显微镜,德国徕卡公司;UV-5100型高性能紫外可见分光光度计,上海让奇仪器科学有限公司;F4500型荧光分光光度计,日本日立公司。
1.3 实验方法
1.3.1大豆亲脂蛋白提取
大豆LP组分的分离由文献[12]的方法经修改后使用。大豆磨粉,过60目筛,正己烷脱脂制备低变性大豆脱脂粉,在70℃干热处理2 h,此时氮溶指数降至75%。50 g干热处理后的脱脂豆粉加入到400 mL蒸馏水中,用NaOH调节pH值到8.0。在20℃下搅拌1 h后3 000g离心10 min。分离上清液并加入 10 mol/L Na2SO3,然后用H2SO4调节pH值至5.8,3 000g离心10 min,再用H2SO4调节上清液pH值至5.0,并在55℃下处理15 min。然后加入50 mol/L NaCl并用NaOH调节pH值到5.5,3 000g离心10 min,沉淀即为LP组分。
1.3.2LP-HPMC复合物的超高压处理
将HPMC与LP以1∶5(质量比)的比例混合于100 mL锥形瓶中,LP质量浓度为5 mg/mL,室温(20℃)搅拌后静置12 h。次日,将混合溶液分成5组,密封后将其中4组放入聚乙烯袋内,置于高压容器中。在25℃条件下,对每组溶液分别进行200、300、400、500 MPa加压处理,每种压力条件下的连续加压时间为10 min。每组测量3次,取平均值,冻干部分备用。
1.3.3LP-HPMC复合物乳液的制备
用pH值为7.4的10 mol/L磷酸盐缓冲溶液制备由HPMC稳定的LP水包油乳液。通过将适量的经不同超高压处理的 LP-HPMC复合物溶液中加入25%的葵花籽油,在10 000 r/min条件下均质3 min后形成乳液,4℃条件下贮藏。
1.3.4三维荧光光谱
采用荧光光谱仪并参照文献[13]的方法测定LP-HPMC溶液的三维荧光光谱,将制备好的LP-HPMC溶液稀释50倍后,取一定量置于比色皿中测定得到三维荧光结构图。初始激发波长为280 nm,扫描波长为200~500 nm,谱带宽度10 nm,扫描16条曲线。
1.3.5红外光谱(FTIR)
将乳液直接进行冻干,冻干后的样品置于干燥器中用P2O5充分干燥,取样品1.5 mg与200 mg溴化钾研磨混匀后压片进行红外光谱测定。在实验过程中,为了减少水蒸气的影响干扰,用干燥的N2持续注入测量室。测定时波数为4 000~400 cm-1,扫描次数64,分辨率4 cm-1,得到的红外吸收曲线采用peak fitting软件和高斯曲线拟合,分析蛋白在不同均质压力的情况下二级结构含量的变化[14]。
1.3.6粒径和电位
使用Mastersizer2000型粒径电位仪进行LP-HPMC溶液粒径分布和ζ-电势的测量。在装入PCS8501型比色杯之前,用10 mol/L磷酸盐缓冲液(pH值7.4)将样品稀释至0.1%。所有测量均在25℃下进行3次。对于分散体使用1.450的折射率,对于连续相(10 mol/L磷酸盐缓冲液,pH值7.4)使用1.331的折射率[15]。
1.3.7溶解性
参照文献[16]的方法稍作修改测定LP溶解性,取冻干粉末溶解于蒸馏水中,质量浓度约为10 mg/mL的蛋白质溶液,搅拌1 h使样品溶解,静置2 min后,将静置后的上层溶液倒入离心管中离心15 min (10 000g)。取上清液2 μL稀释10倍,采用BCA(Bicinchonininc acid)法测定蛋白质含量。以牛清蛋白为标准物,蛋白质溶解度为上清液中蛋白的含量占样品中总的蛋白含量百分比。
1.3.8表面疏水性
参照文献[17]的方法稍作修改,采用ANS(8-苯胺基-1-萘磺酸铵)作为荧光探针测定蛋白质表面疏水性。分别称取 0.025 g不同冻干样品溶于50 mL磷酸盐缓冲液(10 mol/L,pH值7.4)中,在室温条件下搅拌1.0 h后离心(10 000g,30 min),取上清液用BCA法测定蛋白浓度,并用磷酸盐缓冲液(10 mol/L,pH值7.4)依次稀释蛋白,使其质量浓度在0.005~0.1 mg/mL之间,将所得梯度的上清液溶液与8.0 mmol/L ANS以体积比100∶1混合,静置3 min后测其荧光强度。激发波长390 nm,发射波长497 nm,夹缝为5 nm。以荧光强度对蛋白质质量浓度制图,初始段斜率即为蛋白质分子的表面疏水值。
1.3.9乳化活性与乳化稳定性
样品乳化性及乳化稳定性测定参照文献[18]的方法。在0 min和10 min时从乳液样品底部分别取样50 μL,经SDS(十二烷基磺酸钠)稀释200倍,充分混合后在500 nm处测定吸光度,记录为A,以SDS做空白对照。乳化活性指数(Emulsification activity, EAI)和乳化稳定性指数(Emulsification stability, ESI)分别表示为
(1)
(2)
式中E1——乳化活性指数,m2/g
E2——乳化稳定性指数,min
T——活性常数,取2.303
N——稀释倍数,取200
θ——油相体积分数,取25%
L——比色杯厚度,取1 cm
C——乳化液形成前蛋白质水溶液中蛋白质质量浓度,取5 mg/mL
A0、A10——乳状液在0、10 min的吸光度
1.3.10微观结构测定
参照文献[19]的方法稍加修改以用于乳液微观结构观察。用0.01 mol/L pH值7.0的磷酸缓冲溶液将样品适度稀释,取一滴适度稀释的乳液放置在载玻片上,并用盖玻片固定,在40倍光学显微镜下进行观察。
1.4 数据处理
本实验数据均为3个平行样的平均值,结果采用SPSS 22.0分析软件和Origin 8.0软件进行处理,采用ANOVA对数据进行差异显著性分析(P<0.05)。
2 结果与分析
2.1 三维荧光分析
三维荧光是在二维基础上衍生出的新型荧光分析手段,主要用于描述蛋白的荧光强度在两种维度上的变化。它在二维的基础上增加了测量的维数,更加便于直观观测荧光强度状态的变化,增加了光谱的分辨率[20]。
LP和HPMC之间的相互作用很大程度上影响LP的物化性质,从而对LP进一步应用产生影响,可以使用三维荧光分析来研究。图1显示在不同超高压条件处理下LP-HPMC复合物的三维荧光图谱,实验组前期研究证明LP的激发波长和发射波长都在220 nm处,证明其发色基团主要出现在此波长下;当LP与HPMC相互结合时在激发波长280~480 nm范围内,出现第2个峰,证明二者发生相互作用。实验数据显示,不同超高压处理的LP-HPMC复合物荧光强度均高于未经超高压处理的复合物,且当超高压处理条件为400 MPa时,220 nm处荧光强度几乎为零,这可能是由于在此均质压力下LP的发色基团被埋在复合物的疏水区域中,LP荧光强度在复合物的存在下发生猝灭,暂时无法被识别。这也可以从侧面体现400 MPa压力下LP溶解性得到很大提升,增大与HPMC反应量更有利于LP-HPMC复合物的进一步利用。
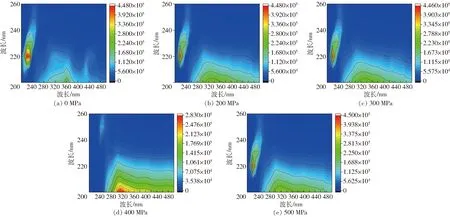
图1 不同超高压处理条件下LP-HPMC复合物三维荧光图谱Fig.1 Three-dimensional fluorescence of LP-HPMC complex under different ultra-high pressure treatment conditions
2.2 红外光谱分析


图2 不同超高压处理条件下LP-HPMC复合物 红外光谱Fig.2 IR spectrum of LP-HPMC composite under different ultra-high pressure treatment conditions
另外,从二级结构数据(表1)分析,超高压处理可以提高α-螺旋相对含量,β-折叠、β-转角和无规则卷曲结构也发生不同程度的改变,400 MPa时α-螺旋、β-折叠结构相对含量最高,说明LP的二级结构在一定程度上被破坏,柔性结构增加,分子由有序结构变得无序。高压均质处理后可能破坏了二级结构中的氢键作用,使蛋白质分子展开、二级结构破坏、蛋白质不同程度改变和伸展,从而影响LP与HPMC的结合程度。
2.3 粒径和电位分析
图3(图中不同字母表示电位差异显著)为不同超高压处理条件形成的LP-HPMC复合物溶液的粒径和电位。粒径越小、越均匀越有利于乳液的稳定;同样电位的绝对值越大表明乳液稳定性越好[24]。粒径图可以看出,未经超高压处理的LP-HPMC溶液出现明显的分峰,超高压处理后的溶液都只有一个粒径峰值,这与二者的结合程度有关,超高压可提高LP与HPMC的结合度。均质压力为400 MPa时,LP-HPMC复合溶液粒径体积分数和平均粒径(218 nm)都最小,同时电位绝对值(16.4 mV)最大,说明此均质压力条件有利于LP-HPMV复合溶液的进一步开发利用。结合共聚焦图和电位变化

表1 不同超高压处理条件下LP-HPMC复合物二级结构相对含量Tab.1 IR spectrum of LP-HPMC composite under different ultra-high pressure treatment conditions
注:同一列不同字母表示有显著性差异(P<0.05)。
可知400 MPa处理的LP与HPMC形成复合物的乳液形状规则稳定,表面净电荷较多,这主要是由于在不同压力下蛋白质的空间构象发生变化,同时HPMC可以改变乳液表面电荷以及增加界面层厚度,增强乳滴间的空间排斥力和亲水性,改善复合物溶液的稳定性[25]。
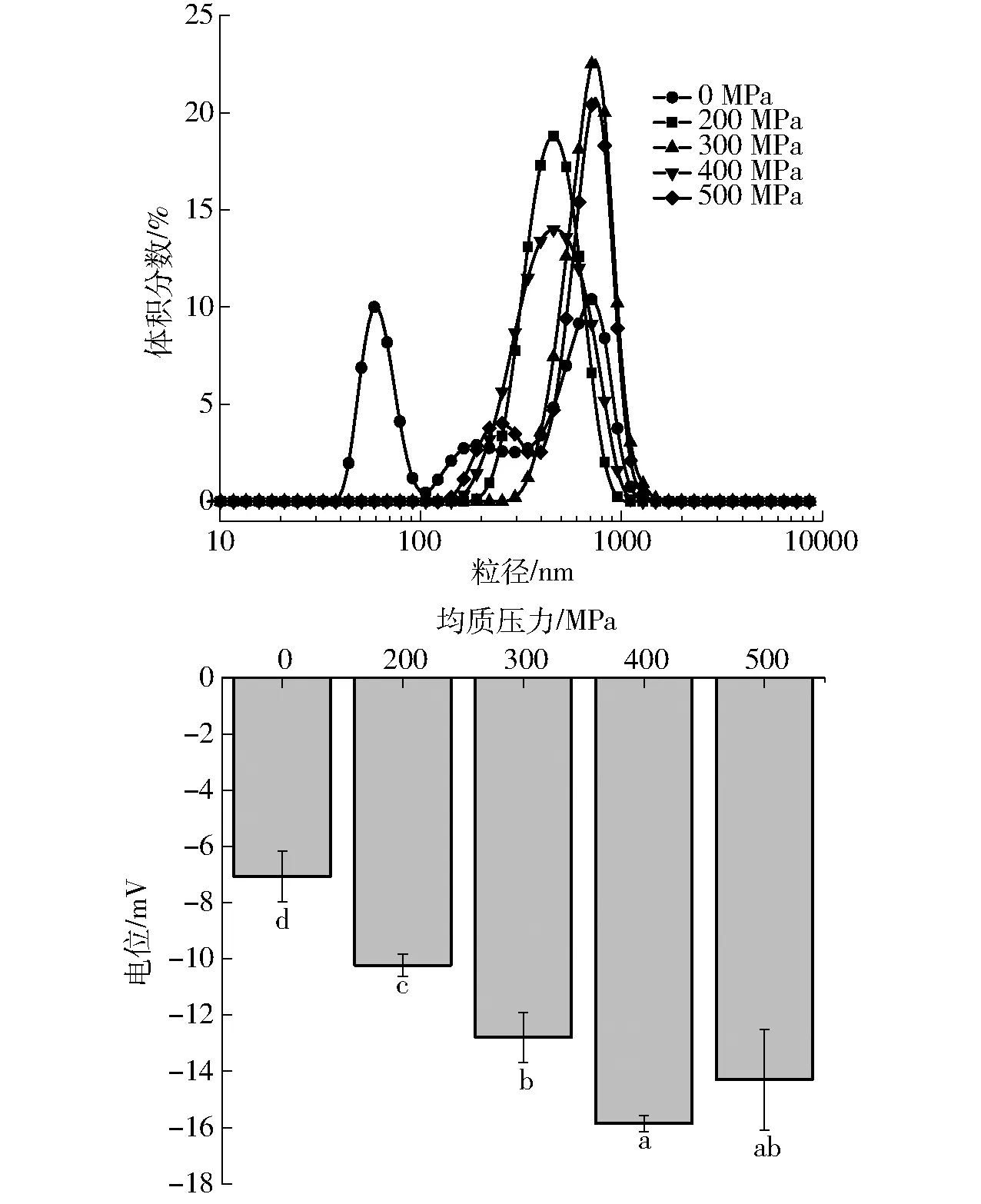
图3 不同超高压处理条件下LP-HPMC复合物 粒径和电位Fig.3 Particle size and potential of LP-HPMC complex under different ultra-high pressure treatment conditions
2.4 溶解性和表面疏水性分析
溶解性是蛋白质的重要指标,可直接影响蛋白质的功能性质[26],而LP由于含大量磷脂,其溶解性较差,HPMC添加可轻微改善LP溶解性,但效果不明显,辅以超高压处理可显著提高LP溶解度,400 MPa时溶解度达到最大值41.1%。在超高压处理过程中发生的均质化效应会产生强大的均质力,这会增加LP-HPMC疏水区域的局部温度和压力,导致蛋白质的去折叠,进而使其内部亲水性氨基酸残基暴露,并且由于超高压处理期间促使LP中的不溶性沉淀物与HPMC相互作用形成可溶性聚集体,因此溶解度得到显著提升。表面疏水值表示蛋白质分子表面上存在的疏水基团数量,蛋白质的疏水值越大溶解度越小。图4(图中不同字母表示溶解度差异显著)可以看出,随着压力的增大,表面疏水值呈现先减小后增大的趋势,与溶解度结果一致,即在400 MPa表面疏水值最小,为57 832,此时溶解度最大。
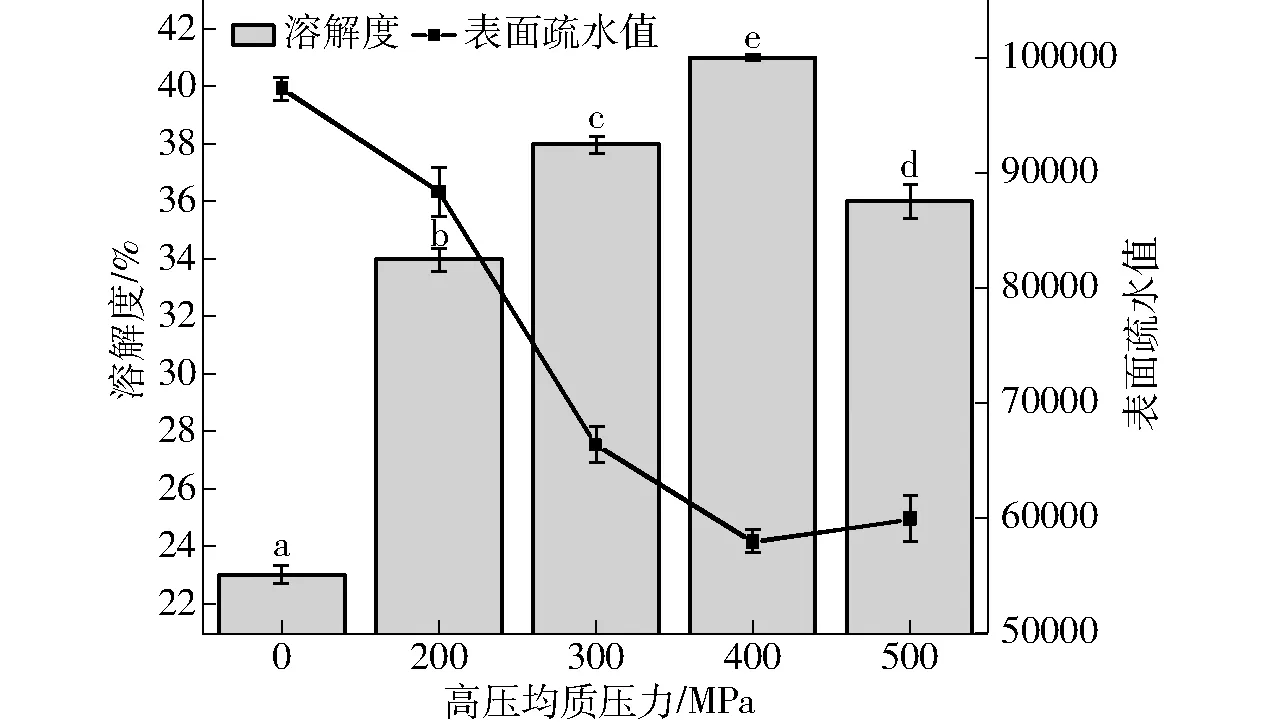
图4 不同超高压处理条件下LP-HPMC复合物 溶解度和表面疏水值Fig.4 Solubility and surface hydrophobicity of LP-HPMC complex under different ultra-high pressure treatment conditions
2.5 乳化活性和乳化稳定性分析
EAI表示的是LP-HPMC复合物形成油-水界面的能力,HPMC结合了LP后在水相中的溶解能力以及HPMC能够强烈吸附在油-水界面形成乳化层的能力;ESI是指乳状液形成小液滴的稳定能力[27]。因此,EAI及ESI是表征LP-HPMC复合物功能性质最有力的指标之一。由图5(图中不同字母表示EAI差异显著)可知,随着均质压力的增大,EAI与ESI都呈现先增大后减小的趋势,在400 MPa均质压力的情况下,LP-HPMC乳液EAI都表现出最大值,超高压力为400 MPa时对于乳液体系最有利,与前文得出的结论一致。而在500 MPa时,EAI与ESI都呈现出最小值,这可能是由于均质压力过高会使LP内部疏水基团暴露,影响其溶解度,从而减少LP-HPMC复合物的形成量,进而使乳液EAI和ESI呈现最小值。这与三维荧光和溶解度数据结果一致,再次证明超高压力为400 MPa时不但有利于LP与HPMC充分结合,此条件形成的乳液乳化活性与乳化稳定性也最佳。
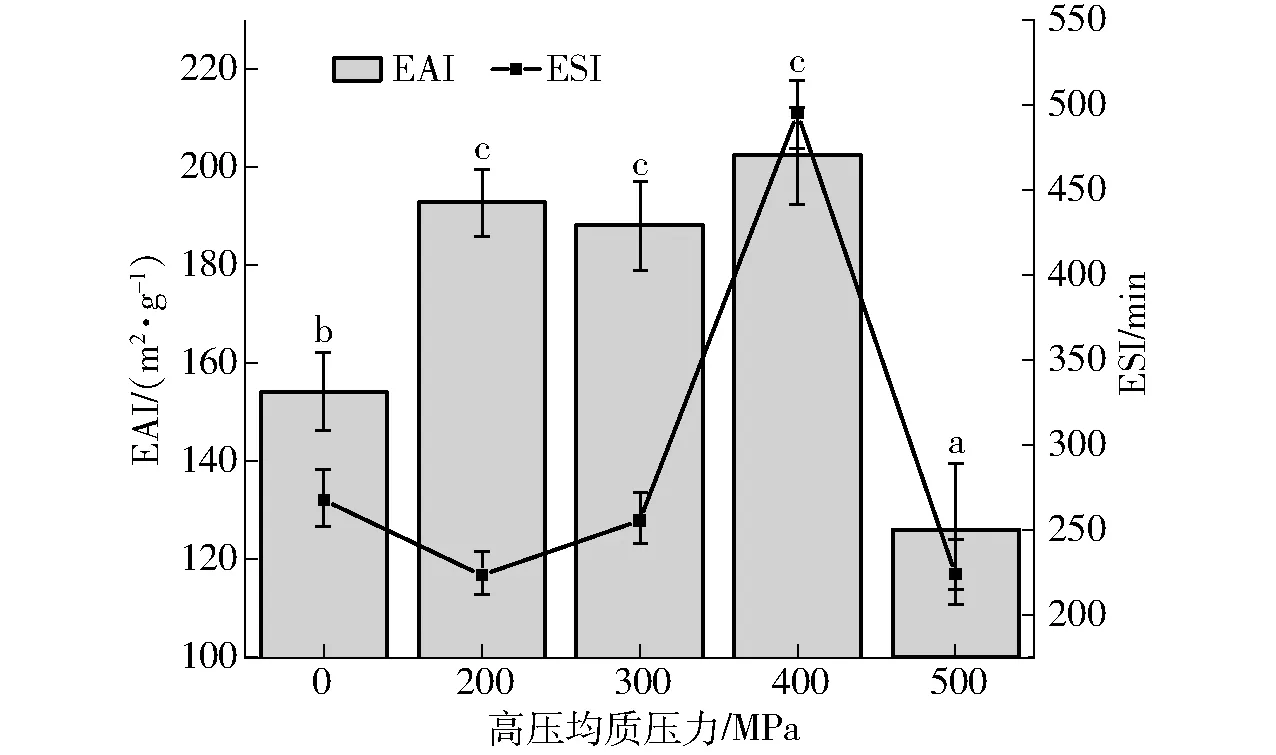
图5 不同超高压处理条件下LP-HPMC复合物 乳液EAI和ESIFig.5 EAI and ESI of LP-HPMC composite emulsion under different ultra-high pressure treatment conditions
2.6 微观结构分析
光学显微镜经常用于分析液体微观结构,能够直观地反映出溶液颗粒大小、分散状况及产生的不稳定现象。图6是不同复合物所形成溶液的微观结构图及平均粒径数据,随着超高压处理压力的增大,溶液粒径呈现先减小后增大的趋势。由图6a可知,未经超高压处理的LP-HPMC溶液液滴粒径较大(1 644 nm)且分布不均匀,出现了液滴聚集,这主要是由于此时LP的溶解性较差,LP和HPMC发生分离,没有较好地复合。LP与HPMC相互作用形成的界面膜不够稳定,易发生絮凝或聚集现象。图6b~6d可以观察到较为均匀的蛋白液滴分布,同时也存在较小的液滴,这主要是由于不同超高压处理的LP与HPMC复合物稳定的液体粒径相对较小且经均质力作用呈球形分布。并且复合物随压力的增加液滴变小、分布均匀。这可能是由于复合物在空气-水界面形成更加致密的膜结构[25-26],LP-HPMC复合物经超高压处理后柔韧性和乳化性较好,形成液滴结构更加均匀,改善了液滴凝聚和絮凝现象,说明超高压处理得到的LP-HPMC复合物能更好地稳定乳液。然而,压力过大依旧不利于稳定复合物的形成,图6e显示500 MPa压力处理下的液滴团聚效应强于200~400 MPa,说明压力过高时反而使蛋白质发生聚集无法伸展,不利于稳定乳液的形成。

图6 不同超高压处理条件下LP-HPMC光学显微结果Fig.6 LP-HPMC optical microscope under different ultra-high pressure treatment conditions
3 结束语
采用超高压处理探究均质压力对LP-HPMC相互作用及复合物功能性质的影响,结果表明:适宜的超高压处理可以显著提高LP与HPMC的复合程度,同时提升功能性质,如溶解性、表面疏水性、乳化活性及乳化稳定性;400 MPa改性的柔性大豆LP与HPMC复合物表现出较好的粒径分布,相对于未经超高压处理的LP-HPMC复合物溶液分散性更好;超高压处理改善了LP二级构象,随着压力的增加,α-螺旋相对含量先升高、后降低,无规卷曲结构相对含量先降低、再升高;400 MPa时,α-螺旋、β-折叠结构相对含量最高,LP构象发生转变,此时LP有序构象的组成、柔性结构的展开影响蛋白质整体构象的柔韧性,更易与HPMC形成功能特性较好的复合物。
猜你喜欢
土壤学报(2022年3期)2022-08-26 12:12:18
原子与分子物理学报(2021年2期)2021-03-29 07:30:46
环境卫生工程(2020年3期)2020-07-27 01:19:22
中成药(2018年7期)2018-08-04 06:04:18
中成药(2018年3期)2018-05-07 13:34:18
环境科技(2016年2期)2016-11-08 12:18:20
中国果菜(2016年9期)2016-03-01 01:28:39
化工进展(2015年6期)2015-11-13 00:32:09
中国洗涤用品工业(2015年11期)2015-02-28 19:03:09
济宁医学院学报(2014年4期)2014-08-16 13:44:19
