
基于一维卷积神经网络的蛋白质ATP绑定位点预测
2019-12-23 07:19张寓於东军
计算机应用 2019年11期
张寓 於东军

摘 要:在生物内部活动中,蛋白质和配体之间的互相作用是非常常见而又重要的一种活动,腺嘌呤核苷三磷酸(ATP)是其中一种非常重要的配体。为了提高预测腺嘌呤核苷三磷酸(ATP)绑定位点的准确率,提出了一种基于一维卷积神经网络(1DCNN)的方法。首先,以蛋白质的序列信息为基础,融合位置特异性得分矩阵信息、二级结构信息和水溶性信息,使用随机下采样的方法消除数据不平衡的影响,再对缺失的特征进行再编码补齐,得到训练特征。训练一个1DCNN来预测蛋白质ATP绑定位点,优化网络结构,并且进行实验来对比所提方法和其他机器学习方法的优劣。实验结果展示了所提方法的有效性,并且该方法与传统支持向量机(SVM)相比在AUC指标上有部分的提升。
关键词:蛋白质ATP;卷积神经网络;数据不平衡问题;分类
中图分类号:TP391.4
文献标志码:A
ProteinATP binding site prediction based on 1Dconvolutional neural network
ZHANG Yu, YU Dongjun*
School of Computer Science and Engineering, Nanjing University of Science and Technology, Nanjing Jiangsu 210094, China
Abstract:
Interaction between proteins and ligands is a very common and important activity in the internal activities of organisms. Adenosine TriPhosphate (ATP) is one of the most important ligands. To improve the accuracy of proteinATP (Adenosine TriPhosphate) binding sites, an algorithm was proposed by using One Dimensional Convolutional Neural Network (1DCNN). Firstly, based on the protein sequence information, position specific score matrix information, secondary structure information and water solubility information were combined and random undersampling was used to eliminate the impact of data imbalance. Then, the missing features were completed by recoding. Finally, the training features were obtained. A 1DCNN was trained to predict proteinATP binding sites, the network structure was optimized, and experiments were carried out to compare the proposed method and other machine learning methods. Experimental results show that the proposed method is effective and can achieve better performance on AUC (Area Under Curve) compared to the traditional Support Vector Machine (SVM).
Key words:
proteinATP (Adenosine TriPhosphate); Convolutional Neural Network (CNN);data imbalance problem;classification
0 引言
腺嘌呤核苷三磷酸(Adenosine TriPhosphate, ATP)是大部分生物体力最直接的能量来源,它水解时能释放出大量的能量,这些能量参与了很多人类的基础的生命活动, 因此,ATP是人体内极为重要的一种分子[1-2]。蛋白质ATP绑定位点指的是在绑定有ATP的蛋白质的残基中和ATP配体距离小于一定距离的残基,如果能够正确定位蛋白质ATP绑定位点,就能变相地确定ATP分子的位置,这对药物设计和进一步的生物生命活動研究都有着重大的意义[3-4]。
在传统的生物学实验中,通常要通过生物湿实验来测定蛋白质的ATP绑定位点,但是做生物湿实验较为耗时而且经济成本高昂。随着数字化信息时代的开始,很多的生物信息以数字化的方式得以保存[5],随后就出现了使用已知的生物信息来预测未知的信息的方法,最开始使用的方法一般是基于模板的匹配算法[6-7],随着机器学习技术的发展,开始出现使用机器学习来预测未知的绑定位点[8-9]的方法。
在机器学习方法中,有效的特征提取是提高预测性能的关键。在蛋白质的特征工程方面,开始只是使用原本的序列信息来预测,后来,蛋白质序列中的一些隐藏的信息被挖掘了出来,例如:位置特异性得分矩阵(Position Specific Score Matrix, PSSM)[10]、二级结构(Secondary Structure, SS)信息、溶液接触面积(Solvent Accessibility, SA)信息、物理化学属性[11]、三级结构信息[12-13]等,这些信息也被用于参与预测,而且提高了预测的性能; 然而这些信息很多也是未经过实验测定的。针对这个问题,后续出现了使用预测器来预测这些隐藏信息,并且把预测结果作为特征参与预测的集成预测器[14-15]。随着方法的不断改进,预测的准确率也越来越高。
近几年来,深度学习技术开始逐渐应用于生物信息学,DeepBind[16]使用了深度卷积神经网络预测了脱氧核糖核酸(DeoxyriboNucleic Acid, DNA)和核糖核酸(RiboNucleic Acid,RNA)的蛋白质绑定位点。DeepSite[17]直接使用蛋白质的三级结构特征训练一个深度三维卷积神经网络,对绑定口袋在蛋白质三维空间中的位置进行预测。随着这些深度学习的方法的产生,预测的准确率又被提高了一个层次。
本文提出了一种基于一维卷积神经网络(One Dimensional Convolutional Neural Network, 1DCNN)的方法来预测蛋白质ATP绑定位点,使用蛋白质的序列信息、PSSM矩阵、二级结构特征、溶液接触面积特征来训练模型,预测每个残基是绑定位点的概率。最后和其他使用支持向量機(Support Vector Machine, SVM)的方法进行比较,分析本文方法的优点和缺点。
1 数据集
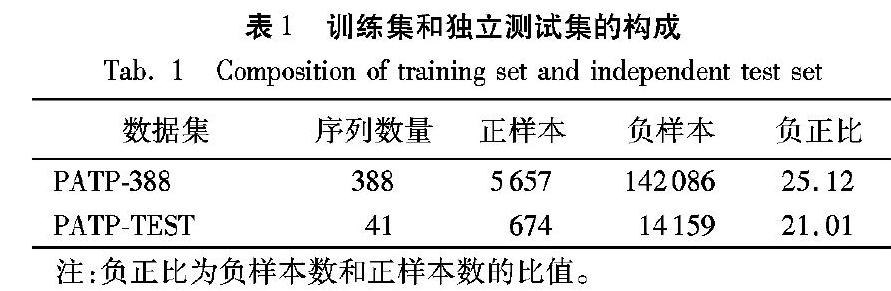
本文所使用的数据集为论文ATPbind[14]中所使用的数据集,该数据集由429个和ATP绑定的蛋白质链构成。该数据集来自2016年5月之前PDB(Protein Data Bank)[18]中的数据,并且使用CDhit[19]软件将同源性超过40%的蛋白质全部除去。在这429个蛋白质链中,将其中388个作为训练集(PATP388),剩下41个蛋白质链作为独立测试集(PATPTEST)。PATP388包含5-657个ATP绑定位点残基和142-086个非ATP绑定位点残基,PATPTEST包含674个ATP绑点位点残基和14-159个非ATP绑点位点残基。
2 蛋白质残基的特征表示
蛋白质的每个残基都是20种氨基酸脱水缩合后中的一种,一条蛋白质链可以简单表示为一个由20种残基组成的字符串,但是每条蛋白质链中的信息远多于一个字符串所表达的信息。蛋白质的特征工程问题一直是生物信息学中复杂而又重要的问题。本文选取了几个常用而又有效的特征,在对其进行一些处理后,作为最终的训练特征。
2.1 特征选取
本文选用了PSSM特异性得分矩阵、蛋白质二级结构特征、溶液接触面积作为基本的特征组成成分。
PSSM矩阵可以反映出该蛋白的序列信息和族谱信息。通常使用PSIBLAST[20]程序将待比对蛋白的信息放到SwissProt[21]蛋白质数据库中搜索并和结果进行比对,最后得到PSSM矩阵。PSSM矩阵是一个N×20的矩阵,N为蛋白质残基的数量,每个残基对应着20个元素,这20个元素代表着20种氨基酸的每一种出现的可信度得分。在得到PSSM矩阵后,使用归一化函数将PSSM矩阵的每个得分进行归一化处理。归一化函数如下:
f(x)=x-minmax-min(1)
蛋白质的二级结构(SS)指的是多个残基在局部所表现出的一种链接形状。蛋白质的二级结构被分为α螺旋(H)、β折叠(E)和无规律的卷曲(C)。本文中用已有的预测器psipred[22]来预测蛋白质的每个残基属于某一种二级结构的概率,并把得到的结果作为特征。预测器得到的结果为N×3的矩阵,即每个残基有3个元素,分别指3种不同二级结构的概率。
蛋白质的溶液接触面积(SA)是指蛋白质每个残基可以和水接触的面积大小,间接反映了这个残基是否可溶于水。每个残基对应着三种状态:易于和水接触、不易于和水接触、中性。本文中使用已有的预测器Sann[23]来预测,得到N×3的矩阵,即每个残基对应3个元素,分别代表3种状态的概率。
融合以上特征,每个残基一共有20+3+3=26维的基本向量。然而,一个ATP绑定位点的确定和邻近的残基有相互作用的关系,所以,一般使用一个滑动窗口把指定残基的邻近残基也作为特征输入,针对ATP绑定位点预测问题的滑动窗口大小一般都取17[24],指定残基的前8个和后8个残基也都在滑动窗口之内。但是一条蛋白质链的前8个残基和后8个残基的滑动窗口区域并不存在17个残基。针对这个问题,本文使用了补位的方法,在头部和尾部补上缺失的残基,对于这些补位的残基,使用了一种类似于onehot编码的方法。针对选取的3种基本特征,本文在这3个基本特征中每个额外加入一个维度也就变成了21(PSSM)、4(SS)、4(SA),一共29个维度。对于正常的残基,这3个额外的维度值都为0,而针对补位的残基,这3个维度值为1而其余的维度值为0。
最后,每个残基的维度数量就变成了29×17=493个,这493个维度也是作为最终的输入向量的构成。
2.2 随机下采样
由于正负样本数量极为不平衡,正负类比达到1∶24。这就造成了不平衡问题[25]。如果不对数据进行处理,直接进行训练,那么分类器将倾向把所有样本都预测为负类。针对不平衡问题,通常会使用上采样或者下采样方法来平衡样本。文中所使用的是最常用的随机下采样,即随机选取指定数目的负类样本,使之数目和正类样本平衡。每次随机拿取正类样本数目的负类样本,和所有正类样本混合作为一轮神经网络训练的训练样本。
3 卷积神经网络模型的构建和训练
3.1 网络模型结构
由于蛋白质序列信息是一维信息而且具有局部关联性,本文使用一维卷积神经网络作为模型进行训练。神经网络的大致结构如图1所示。神经网络的输入是一个17×29的矩阵,即长度为17,深度为29。
网络的第一层和第二层都为卷积层(Conv Layer),卷积的窗口大小为2,步长为1,第一层卷积核的数量为400,第二层卷积核的数量也为400,所以经过2层卷积之后,输出的数据的深度就为400。
由于序列所携带的信息量比较少,在图像领域中的卷积神经网络中常用的池化层在本文中并没有使用。
第三层为平铺层(Flatten Layer),用来连接卷积层和全连接层。
第四层和第五层都为全连接层(Dense Layer),第四层的神经元的数量为500,第五层为200。
第六层是带有丢弃[26](Drop out)的全连接层,随机丢弃一些输入来防止神经网络过拟合,本文中丢弃输入的概率设为50%。
最后一层为输出层(Output),神经元的数量为2。这两个神经元的输出就分别代表着正类和负类的预测置信度。
在每一层得到输入计算出结果后,会使用激活函数对结果进行调整。除最后输出层外,每一层都使用了ELU激活函数。第五层则使用了sigmoid函数。ELU和sigmoid函数如下:
elu(x)=x, x≥0α(exp(x)-1),x<0 (2)
sig(x)=11+e-x(3)
最后对这两个输出结果使用Softmax函数,得出正类和负类的预测概率。Softmax函数如下:
σ(z)j=ezj∑Kk=1ezk(4)
本次实验使用TensorFlow[27]进行神经网络的构建和训练,在硬件上使用了两块GTX Titan XP,并且使用CUDA(Compute Unified Device Architecture)运算平台进行加速计算。
为了优化网络结构,本文还构建了一些不同结构的神经网络进行对比实验。文中主要通过改变网络模型中卷积层的层数和每层卷积层中卷积核的个数来改变结构。层数越多,卷积核数量越多的网络的学习能力也就越强,但是同时也会导致学习的速度慢、梯度爆炸和梯度消失的情况发生。
3.2 模型训练
本文将一次随机下采样的样本作为一轮的训练样本进行训练,将每次Softmax函数得到的正类和负类的预测概率和真实类别的交叉熵函数作为损失函数,使用梯度下降法对网络模型进行优化。交叉熵函数如下:
H(p,q)=∑ip(i)*ln(1/q(i))(5)
其中:p为样本的真实值; q为样本的预测值; p(i)、q(i)表示向量p、q的第i个元素。
为了使模型收敛的速度加快,将正负样本交替输入进模型进行训练,每输入一个残基的特征就进行一次梯度下降操作,即batch size=1。本文以对独立测试集预测结果的AUC(Area Under Curve)作为第一评估指标,训练多轮直至AUC收敛。
4 实验结果和比较分析
4.1 评判标准
本文涉及的是一个二分类问题,所有样本只有正类和负类。根据独立测试集的真实标签和预测标签,可以把每个样本的预测结果分成4种情况:TP(True Positives)、FP(False Positives)、TN(True Negatives)和FN(False Negatives)。
根据这些基础数据,演化出了特异性(Specificity,Spe)、敏感性(Sensitivity,Sen)、准确性(Accuracy,Acc)、马修斯相关系数(Matthews Correlation Coefficient,MCC)。由于本文的分类器是软分类器,即输出的是属于正类的概率,必须先确定一个阈值才能确定预测的样本是正类还是负类,如果得到的正类概率大于阈值那么这个样本就会被预测为正类; 反之,就会被预测为负类。所以,一个已经被预测好的结果的TP、FP、FN、TN是会随着阈值而改变的,MCC等数据也会随之改变。
Spe=TNTN+FP(6)
Sen=TPTP+FN(7)
Acc=TP+TNTP+TN+FP+FN(8)
MCC=
TP*TN-FP*FN(TP+FP)*(TP+FN)*(TN+FP)*(TN+FN)(9)
AUC可以更加稳定地去描述一个预测结果的好坏,将阈值从0到100%遍历,根据TP/TP+FN(纵坐标)和FP/FP+TN(横坐标),画出ROC(Receiver Operating Characteristic)曲线,AUC为ROC曲线下与坐标轴围成的面积,而这个值是不需要一个确定的阈值的。
由于本文所涉及的问题是个样本不平衡问题,如果使用Acc作为衡量标准是无法判别预测器好坏的,因为即使把所有类都预测为多数类,Acc也能达到很高的数值, 所以,一般用AUC指标作为分类器的评判标准。
4.2 实验结果
本文将随机下采样后得到的一组数据作为一轮训练的所需数据。针对每种结构不同的神经网络,会动态调正学习速率,并且让其训练多轮。在每轮训练结束后,使用独立测试集对当前的网络进行一次测试。前170轮次在独立测试集上的结果如图2所示。
可以看出,大概第50轮次训练开始,AUC指数就已经收敛并且趋向于稳定。但是,从40轮开始,MCC却以非常慢的速度上升,而且60轮往后的MCC表现得非常不稳定。随着损失的不断减小,AUC和MCC并没有出现下降的趋势,说明并没有发生过拟合的现象。
可以得出,卷积神经网络模型在本文的问题中对AUC指标的优化是非常有效的,但是对MCC指标的优化却有所不足,而且从50轮开始,AUC数值基本稳定,但是MCC却缓慢提高。MCC指标优化缓慢的问题有可能和本文神经网络所使用的交叉熵损失函数核梯度下降的优化方法有关。
将这种方法在独立测试集上,和其他方法进行比较,结果如表3所示。
从表3可以看出:虽然Acc并不是特别优秀,不及ATPseq的99.27%,但也达到了一般预测器96%的水平;而本文所提出的方法较于其他方法在AUC上有不少提升,比最好的ATPseq也要高出0.4個百分点,达到了88.2%。这可能就是神经网络着重于AUC的优化才导致了这一结果。
5 討论
5.1 一维卷积神经网络还是二维卷积神经网络
二维卷积神经网络目前被广泛应用于图像领域,而且取得了很好的成果。然而,在本文所讨论的问题中,我们认为并没有必要使用二维卷积神经网络。
本文所使用的序列信息是一个一维的信息,虽然产生的PSSM、SS、SA矩阵看似是个二维的信息,但是本文中只有残基之间是有顺序关系的,也就是矩阵中的各个横列是顺序关系的。而矩阵中的纵列只是残基在各个指标上的得分,各个指标之间并没有明显的顺序关系。
本文使用了规模大体一致的一维卷积神经网络(1DCNN)和二维卷积神经网络(Two Dimensional Convolutional Neural Network, 2DCNN)进行训练,并且对比了它们的训练过程中在独立测试集上的结果,如图3所示。
一维卷积网络使用的是2层卷积层、400卷积核的结构。二维卷积网络的卷积窗口大小为2×2,因此卷积窗口的数量就变多了。为了保证网络大小规模的平等性,所以削减了卷积核的数目,为2层卷积层、20个卷积核的结构。
通过实验可以看出,在40轮训练前,二维网络的AUC收敛地稍微快一些。在40轮训练后,两个网络在AUC指标上基本就没有什么明显的差距,而且一维的网络比二维的网络更为稳定。因此,针对序列信息的预测方面,二维卷积网络和一维卷积网络并没有明显的差距。
5.2 深度卷积神经网络还是普通卷积神经网络
一般来说,深度神经网络的学习能力和泛化能力都强于浅层的神经网络,但是针对本文中的序列信息来说,浅层的神经网络已经足够学习到里面的规律。在DNA蛋白质绑定位点的预测问题上,有学者使用了只有1层卷积层的卷积神经网络[30],就完成了对绑点位点较好的预测。
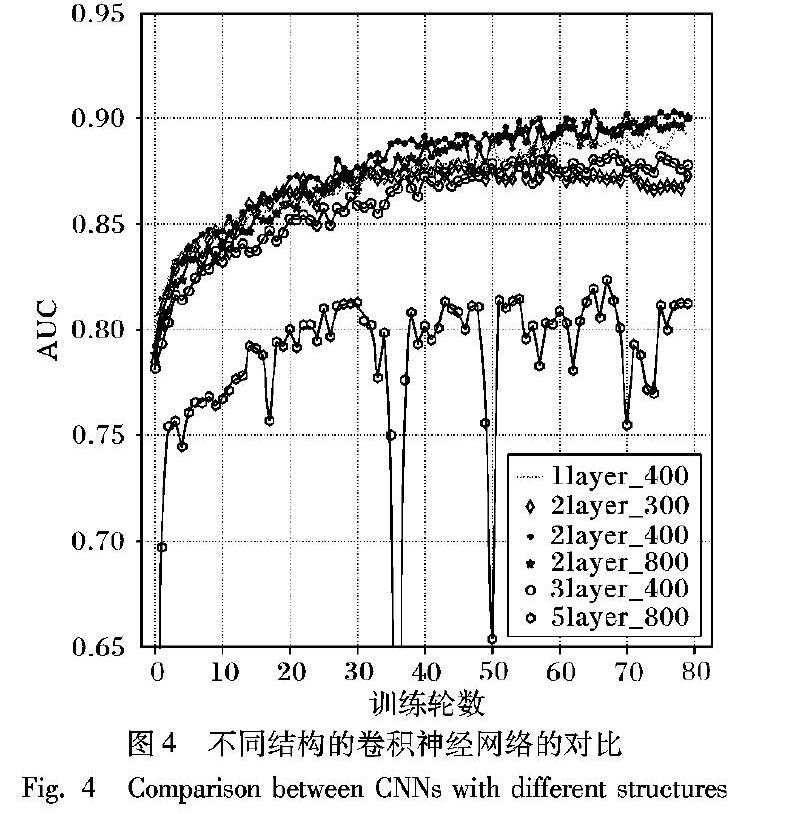
为此,本文使用了3折交叉验证比较了不同规格的卷积神经网络,其结果如图4所示(1layer_400表示1层卷积层400卷积核)。
可以看出,2层400卷积核的神经网络模型已经有很好的学习能力和学习速率,较深网络可能出现了梯度爆炸的问题导致收敛困难,而且在卷积神经网络的深度和神经元数量达到一定规模时,再增加深度或者神经元数量并不会对最终结果产生很大的改变。
6 结语
本文主要研究了蛋白质ATP绑定位点的预测问题,使用了蛋白质的特异性得分矩阵、二级结构、溶液接触面积的融合特征,基于一维卷积神经网络的算法,并且采用随机下采样解决了不平衡问题,完成了对蛋白质ATP绑定位点的较好的预测,并且在独立测试集上得到了验证。
参考文献 (References)
[1]GAO M, SKOLNICK J. The distribution of ligandbinding pockets around proteinprotein interfaces suggests a general mechanism for pocket formation[J]. Proceedings of the National Academy of Sciences of the United States of America, 2012, 109(10): 3784-3789.
[2]TURTON D A, SENN H M, HARWOOD T, et al. Terahertz underdamped vibrational motion governs proteinligand binding in solution [EB/OL]. [2018-11-20].http://europacat.co.uk/staff/wynne/pubs/r/2014NatCommproteins.pdf.
[3]SIRIMULLA S,BAILEY J B, VEGESNA R,et al. Halogen interactions in proteinligand complexes: implications of halogen bonding for rational drug design [J]. Journal of Chemical Information and Modeling, 2013, 53(11): 2781-2791.
[4]AMARI S, AIZAWA M, ZHANG J, et al. VISCANA: visualized cluster analysis of proteinligand interaction based on the ab initio fragment molecular orbital method for virtual ligand screening[J]. Journal of Chemical Information and Modeling, 2006, 46(1): 221-230.
[5]BILOFSKY H S,BURKS C. The GenBank genetic sequence data bank[J]. Nucleic Acids Research, 1988, 16(5): 1861-1863.
[6]LEVITT D G, BANASZAK L J. POCKET: a computer graphics method for identifying and displaying protein cavities and their surrounding amino acids[J]. Journal of Molecular Graphics, 1992, 10(4): 229-234.
[7]LASKOWSKI R A. SURFNET: a program for visualizing molecular surfaces, cavities, and intermolecular interactions[J]. Journal of Molecular Graphics, 1995, 13(5): 323-330.
[8]CHEN K, MIZIANTY M J, KURGAN L. ATPsite: sequencebased prediction of ATPbinding residues[J]. Proteome Science, 2011, 9(S1): S4.
[9]石大宏. 基于序列的蛋白质—核苷酸绑定位点预测研究[D]. 南京:南京理工大学, 2015.(SHI D H. Sequential proteinGDP binding residues prediction[D]. Nanjing: Nanjing University of Science and Technology, 2015.)
[10]JONES D T, WARD J J. Prediction of disordered regions in proteins from position specific score matrices[J]. ProteinsStructure Function and Bioinformatics, 2003, 53(S6):573-578.
[11]LAURIE A T R, JACKSON R M. QSiteFinder: an energybased method for the prediction of proteinligand binding sites [J]. Bioinformatics, 2005, 21(9): 1908-1916.
[12]ZHANG Y, SKOLNICK J. TMalign: a protein structure alignment algorithm based on the TMscore[J]. Nucleic Acids Research, 2005, 33(7): 2302-2309.
[13]楊骥.基于序列与结构特征结合的蛋白质与DNA绑定位点预测[J]. 计算机与现代化, 2016(1):20-25.(YANG J. Prediction of DNAprotein binding sites based on combining sequence with structure information[J]. Computer and Modernization, 2016(1):20-25.)
[14]於东军,胡俊,於铉.基于查询驱动的蛋白质配体绑定位点预测方法: 201310573950.9[P]. 2014-03-05.(YU D J,HU J,YU X. Prediction of proteinligand binding sites based on querydriven: 201310573950.9[P]. 2014-03-05.)
[15]HU J, LI Y, ZHANG Y, et al. ATPbind: accurate proteinATP binding site prediction by combining sequenceprofiling and structurebased comparisons[J]. Journal of Chemical Information and Modeling, 2018, 58(2): 501-510.
[16]ALIPANAHI B, DELONG A, WEIRAUCH M T, et al. Predicting the sequence specificities of DNA and RNAbinding proteins by deep learning [J]. Nature Biotechnology, 2015, 33(8): 831-838.
[17]JIMENEZ J, DOERR S, MARTINEZROSELL G, et al. DeepSite: proteinbinding site predictor using 3Dconvolutional neural networks [J]. Bioinformatics, 2017, 33(19): 3036-3042.
[18]DESHPANDE N, ADDESS K J, BLUHM W F, et al. The RCSB protein data bank: a redesigned query system and relational database based on the mmCIF schema[J]. Nucleic Acids Research, 2005, 33(S1): D233-D237.
[19]LI W, GODZIK A. Cdhit: a fast program for clustering and comparing large sets of protein or nucleotide sequences[J]. Bioinformatics, 2006, 22(13): 1658-1659.
[20]SCHAFFER A A, ARAVIND L, MADDEN T L, et al. Improving the accuracy of PSIBLAST protein database searches with compositionbased statistics and other refinements[J]. Nucleic Acids Research, 2001, 29(14): 2994-3005.
[21]BAIROCH A, APWEILER R. The SWISSPROT protein sequence database and its supplement TrEMBL in 2000[J]. Nucleic Acids Research, 2000, 28(1): 45-48.
[22]JONES D T. Protein secondary structure prediction based on positionspecific scoring matrices[J]. Journal of Molecular Biology, 1999, 292(2): 195-202.
[23]JOO K, LEE S J, LEE J. Sann: solvent accessibility prediction of proteins by nearest neighbor method[J]. Proteins, 2012, 80(7): 1791-1797.
[24]YU D J, HU J, TANG Z M, et al. Improving proteinATP binding residues prediction by boosting SVMs with random undersampling [J]. Neurocomputing, 2013, 104:180-190.
[25]COHEN G, HILARIO M, SAX H, et al. Learning from imbalanced data in surveillance of nosocomial infection [J]. Artificial Intelligence in Medicine, 2006, 37(1): 7-18.
[26]SRIVASTAVA N, HINTON G, KRIZHEVSKY A, et al. Dropout: a simple way to prevent neural networks from overfitting[J]. The Journal of Machine Learning Research, 2014, 15(1):1929-1958.
[27]ABADI M. TensorFlow: learning functions at scale[J]. ACM SIGPLAN Notices, 2016, 51(9): 1.
[28]CHEN K, MIZIANTY M J, KURGAN L. Prediction and analysis of nucleotidebinding residues using sequence and sequencederived structural descriptors [J]. Bioinformatics, 2012, 28(3): 331-341.
[29]YU D J, HU J, HUANG Y, et al. TargetATPsite: a templatefree method for ATPbinding sites prediction with residue evolution image sparse representation and classifier ensemble [J]. Journal of Computational Chemistry, 2013, 34(11): 974-985.
[30]ZENG H, EDWARDS M D, LIU G, et al. Convolutional neural network architectures for predicting DNAprotein binding[J]. Bioinformatics, 2016, 32(12): i121-i127.
This work is partially supported by the National Natural Science Foundation of China (61772273, 61373062).
ZHANG Yu, born in 1995, M. S. candidate. His research interests include bioinformatics computing, pattern recognition.
YU Dongjun, born in 1975, Ph. D., professor. His research interests include bioinformatics computing, machine learning, pattern recognition, intelligence system.
猜你喜欢
大众健康(2021年6期)2021-06-08
东方少年·布老虎画刊(2020年4期)2020-06-08
小天使·一年级语数英综合(2017年11期)2017-12-05
初中生世界·七年级(2017年9期)2017-10-13
少儿科学周刊·儿童版(2017年3期)2017-06-29
科技创新与应用(2016年35期)2017-02-21
计算机应用(2016年12期)2017-01-13
软件导刊(2016年9期)2016-11-07
软件工程(2016年8期)2016-10-25
电脑知识与技术(2016年10期)2016-06-16
