
纳米氧化铪的制备与热膨胀性
2019-11-08 01:45:34刘占宁邢献然
无机化学学报 2019年11期
孙 静 李 强 林 鲲 刘占宁 邢献然*,
(1北京材料基因工程高精尖创新中心,北京 100083)(2北京科技大学固体化学研究所,北京 100083)
0 引 言
氧化铪(HfO2)作为一种高介电常数和宽带隙的材料,由于具有适宜的能带偏移和优良的界面性能,成为可用于取代晶体管中SiO2的栅极绝缘层的替换材料[1]。氧化铪基薄膜从20世纪90年代至今一直是信息技术相关领域研究的重点[2-4]。例如在集成电路中金属-氧化物-半导体场效应晶体管(MOSFET)的栅介质[5-6],以及动态随机存储器(DRAM)的电容介质等[7]。
随着微电子器件的不断微型化和集成化,其尺寸在不断减小,体相材料已经无法满足人们的要求。由于晶体管特征尺寸的迅速缩小,SiO2受摩尔定律的限制漏电流急剧增大,急需高k电介质以取代栅极相关应用中的SiO2[8]。并且自从纳米氧化铪于2008年第一次被英特尔公司用于45 nm处理器金属-氧化物-半导体场效应晶体管栅介质后,氧化铪基纳米材料的商业应用前景也充满活力。
我们知道电子元器件在服役环境内,环境温度会出现较大差异。同时器件自身工作产热也会造成器件局部的强烈热振,电子器件温度交替带来的热膨胀作用会使器件不同部分产生变形,当器件组元的热膨胀系数相差较大时,甚至可能有开裂或者组部件分离的现象发生。如何避免热循环失效是器件应用过程中需要重点关注的问题。
本文通过变温同步辐射X射线粉末衍射和X射线原子对分布函数(pair distribution function,PDF)研究了氧化铪纳米颗粒的晶体结构及在可能的服役温度范围内的热膨胀行为。与体相比较,纳米化的HfO2颗粒由于表面应力的原因晶体结构发生一定程度的畸变,进而改变了晶格的热膨胀行为。对于纳米化HfO2颗粒晶格热膨胀的深入研究及其局域结构信息的揭示,将为HfO2基电子元器件的设计提供重要的结构基础。
1 实验部分
1.1 试 剂
试 剂 HfCl4(98%),乙 醇 (99%), 苯 甲 醇 (98%),KOH(98%),HfO2(98%)均购自 Alfa Aesar。
1.2 样品制备
HfO2纳米颗粒是根据Pinna[9]的方法进行改进而合成的。将1 g HfCl4加入225 mL苯甲醇中,搅拌50 min使其成为均匀的悬浊液,然后加入到500 mL聚四氟乙烯水热釜中,将容器置于恒温鼓风干燥箱中在220℃反应48 h。待自然冷却后,采用乙醇离心清洗3~5次除去苯甲醇溶剂。洗净的纳米粉体在干燥箱中80℃下加热3 h干燥,干燥后的样品放入马弗炉中在500℃煅烧1 h,除去表面的有机物。对于体相HfO2,将样品放入马弗炉中在1 000℃煅烧5 h,使颗粒充分长大。
1.3 仪器与表征
采用日本JEOL JEM-2010透射电子显微镜 (加速电压200 kV)观察样品形貌和结构。样品制样方法为:取少量HfO2纳米颗粒分散在乙醇中,超声3 min后取分散有HfO2纳米颗粒的乙醇溶液滴于覆有碳膜的铜网上,测试中使用明场像模式。采用美国Varian Excalibur 3100傅里叶变换红外光谱仪采集样品的红外光谱。合成样品的相结构通过X射线粉末衍射确定,在日本Spring-8同步辐射光源的BL44B2线站上采集,X射线能量是24.75 keV,X射线波长为0.050 0 nm,光子通量是1011s-1,单色器是Si(111),使用探测器是 MYTHEN DECTRISLtd,角度分辨率为 0.005°,角度采集范围为 5°~150°。 X 射线原子对分布函数(PDF)在美国阿贡国家实验室先进光源(Advanced Photon Source,APS)的11-ID-B线站上采集获取,X射线能量是58.66 keV,X射线波长为0.0211 nm,光子通量是2.3×1012s-1,单色器是Si(311),使用探测器是Varex 4343CT,约化结构函数的傅里叶变换采用PDFgetX3软件获得,倒空间Q值取到2.5 nm-1,进一步数据分析采用PDFgui软件进行。
2 结果与讨论
2.1 形貌和尺寸分析

图1 HfO2纳米颗粒的TEM图片Fig.1 TEM images of HfO2nanoparticles
图1 是HfO2纳米颗粒的透射电镜(TEM)图。图1(a)是大视野的图片,图1(b)是局部照片。从图中可知,HfO2纳米颗粒的尺寸分布比较均一,约为4 nm,但是并不具有规整的形状。这是由于在合成过程中爆发式的形核发生后纳米晶的生长受到反应物浓度大幅降低的限制,纳米晶很难获得同化学成键相匹配的生长速度,从而使其形貌不具有规整的形状[9]。该结果其实是纳米晶周围的动力学条件所决定的。图1(b)中的插图是HfO2纳米晶的高分辨透射图片(HRTEM),从图上的清晰晶格条纹可知尽管HfO2纳米颗粒的粒径很小但依然结晶性很好。
2.2 相成分分析
HfO2在常温至1 700℃温度范围内具有稳定的单斜相[10]。图2是合成的样品的高分辨同步辐射X射线粉末衍射图 (synchrotron X-ray powder diffraction,SPD),对比标准卡片(ICSD No.27313)可以看出HfO2样品均为单斜相。纳米样品由于尺寸效应的原因,衍射峰具有本征的尺寸宽化和重叠。同时比较相对峰强和峰的宽化程度可以发现,各个衍射峰的宽化很一致,各向异性宽和择优取向可以忽略。
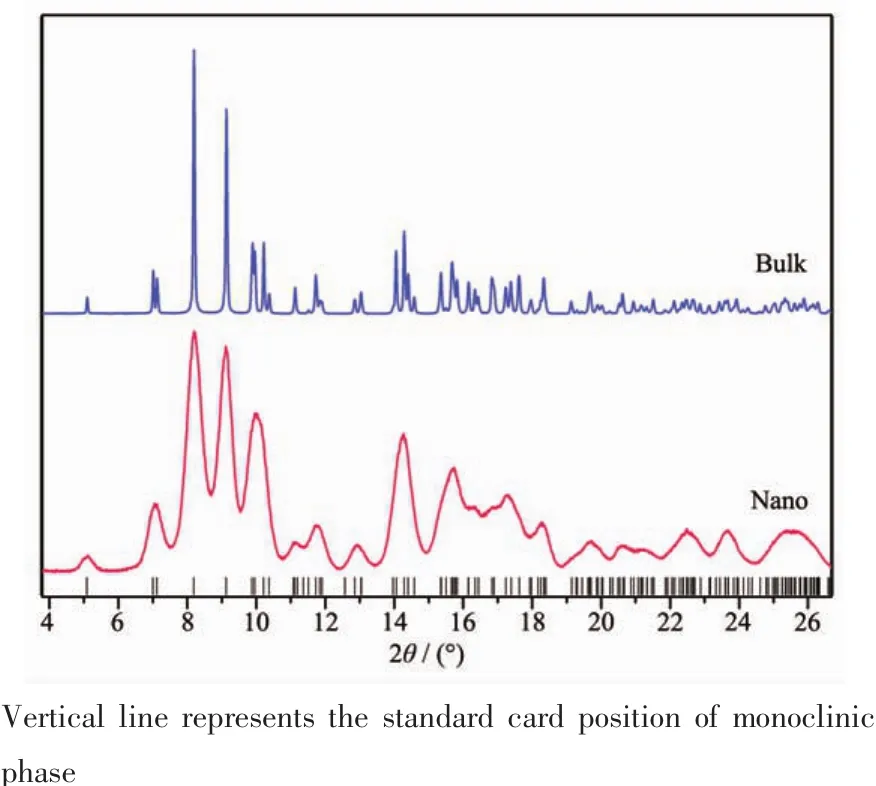
图2 HfO2样品的SPD图Fig.2 SPD spectra of HfO2 sample
2.3 表面状态分析
在纳米颗粒合成过程中,表面包覆剂与颗粒表面容易产生化学键合作用,因而会影响纳米颗粒的物理化学性能。为了消除表面包覆剂对样品的影响,我们通过氧气气氛下烧结使有机物质充分挥发氧化来获得表面干净的颗粒。利用傅里叶变换红外光谱可以对氧化处理前后的样品表面包覆剂是否除尽进行确认。如图3所示,对于样品处理前,3 433 cm-1是水分子的吸收峰,3 022 cm-1是=C-H的伸缩振动峰;2 925和2 850 cm-1对应于-CH2的对称和不对称伸缩振动;2 348 cm-1是空气中CO2的吸收峰;1 613 cm-1对应于OH-的伸缩振动;1 496和1 448 cm-1则分别对应于-COO-的对称和不对称伸缩振动[11]。而分析样品处理后的谱图,我们可以看到属于含有机基团的振动峰均消失了,只剩下水分子、CO2以及OH-1的振动峰,这些物质均包含于环境中。所以由红外光谱分析我们可以认定烧结处理后的样品是表面干净的,其晶格的热膨胀性质是本征的性质,并没有受到表面有机物的干扰。
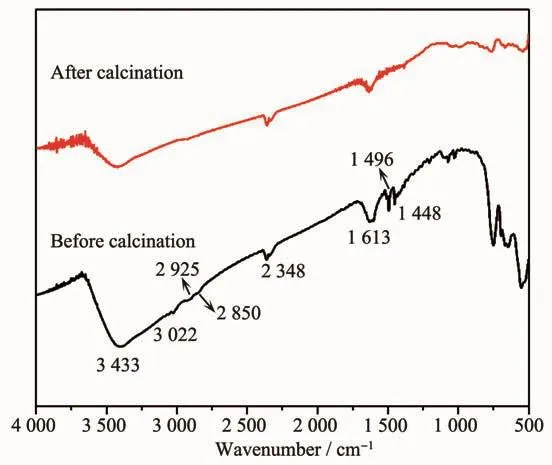
图3 烧结前后HfO2纳米颗粒的红外图谱Fig.3 FT-IR spectra of HfO2 nanoparticles before and after calcination
2.4 热膨胀性能
为了得到HfO2样品的热膨胀数据,需要获得不同温度下HfO2样品的晶格参数。然而,纳米HfO2颗粒中由于晶体长程周期性被破坏,衍射峰具有显著的宽化,甚至是高角度峰的高度重叠(图2),这样使得Rietveld精修过程中对峰位置的判断以及峰面积的计算出现难以避免的系统误差,进而会引起原子坐标、温度因子等提取结果的错误。而X射线全散射技术结合了全局周期性平均结构的布拉格衍射和局域结构的弹性漫散射两种信号,从而弥补了布拉格衍射在局域尺度上信息的缺失[12]。采用X射线全散射信息提取出原子对分布函数可获取更详细而准确的纳米材料的结构信息。所以,HfO2样品的晶格参数通过对X射线原子对分布函数数据的精修来获得。
图4是HfO2体相和纳米样品的PDF谱。由图可知,相比于晶体周期性完美的体相样品,纳米样品的PDF信号衰减地很快,在4 nm左右已经几乎衰减到零。这与纳米样品的尺寸是密切相关的,由于纳米颗粒尺寸约4 nm,原子对分布函数在每个纳米颗粒外的一致性降低,因而PDF峰强在实空间沿径向衰减较快。尽管峰强差异很大,峰的位置与峰型依然较一致,这也说明了纳米和体相样品相结构一致,均为单斜相,没有出现相变。

图4 HfO2样品PDF的拟合结果Fig.4 Results PDF refinement for HfO2 samples
利用PDFgui[13]对实空间4 nm以下的原子对分布函数进行晶体结构的拟合。拟合时采用的初始结构是单斜相(ICSD No.27313),空间群是P21/c。从拟合的结果来看,纳米样品的Rw值为0.11,体相样品的Rw值为0.08,计算与实验数据吻合程度均很好,纳米和体相HfO2的晶体结构可以很好地用单斜相来描述,这点从图4中Gdiff曲线也可以看出。
通过拟合原位变温PDF数据,两组样品的本征热膨胀性能被提取出来,见表1。测试温度范围是-100到200℃,涵盖了HfO2作为电子元器件材料服役温度的极限情况。从表1中数据可知,随着颗粒尺寸的减小,HfO2的单胞参数中a、c收缩,β角减小而b增大,导致单胞呈现一个趋向高温稳定相四方相的变化。已有实验证明,当材料尺寸足够小时,由于表面应力的增加使得材料的高温相在低温时能稳定存在[14]。因此该变化可认为是由于尺寸减小后表面应力的增加造成的。体相HfO2的晶格热膨胀显示出较强的各向异性[15-16],a、c轴随温度增大而呈现出正膨胀行为,b轴正膨胀较小。纳米化后其各向异性的特性增强,从表2数据可知,a、c轴的热膨胀性随晶粒尺寸的减小而增大,b轴热膨胀性稍微减小,最后导致体积的热膨胀性增大,如图5所示。
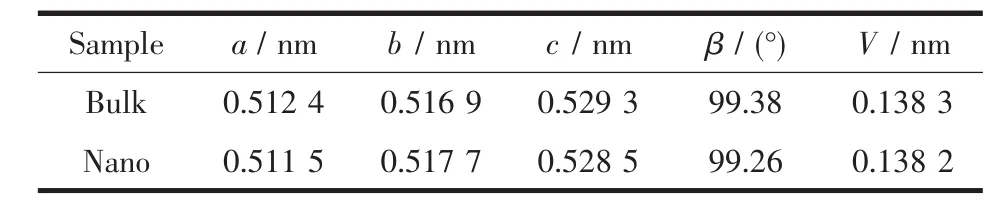
表1 HfO2样品的室温晶格常数Table 1 Lattice constants of HfO2 samples at RT
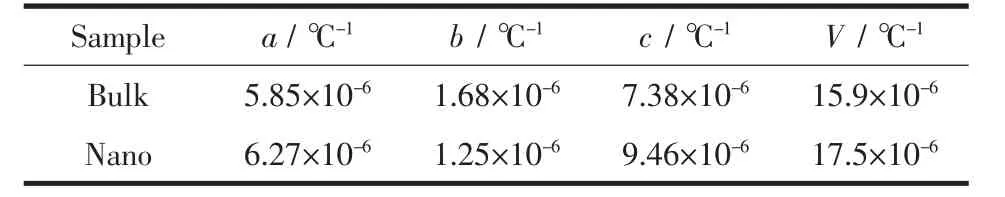
表2 Hf O2样品的热膨胀系数Table 2 Coefficients of thermal expansion of HfO 2 samples
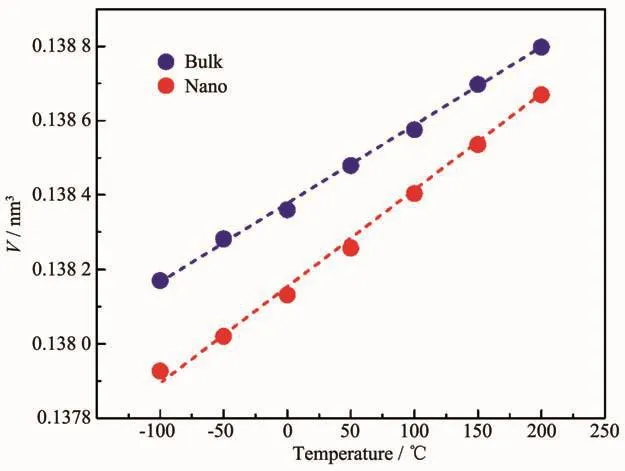
图5 HfO2样品的单胞体积随温度的变化Fig.5 Temperature dependence of HfO2 cell volume
为了更好地理解HfO2尺寸减小后热膨胀性的变化,其详细结构信息通过PDF被提取出来。主要变化源自于通过顶点连接的2个HfO7多面体之间的Hf-O-Hf键角的改变。如图6所示,纳米化后多面体延b轴逆时针旋转,使得Hf-O-Hf键角从147.5°减小至145.4°,该变化可以在PDF(如图7)中Hf-Hf原子对分布函数中观察到,图中左边的峰对应Hf-Hf最近邻原子对,体相和纳米样品的峰位几乎不变,这说明通过边对边连接的HfO7多面体约束性较大,不易受尺寸的影响。右边的峰对应于Hf-Hf次近邻原子对,即通过顶点连接的HfO7多面体中的Hf原子间距。纳米化后该峰左移,即Hf-Hf次近邻原子对距离减小,也验证了Hf-O-Hf键角减小的情况。
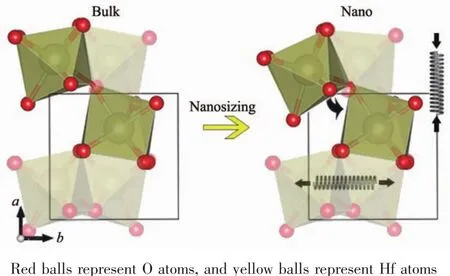
图6 HfO2结构变化示意图Fig.6 Schematic of evolution on crystal structure of HfO2
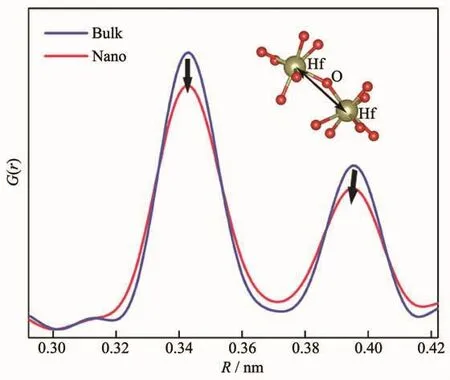
图7 HfO2样品的PDF图谱Fig.7 PDF spectra of HfO2 samples
键角的减小使单胞在a、c轴方向上被压缩,b轴方向上被拉伸。这一系列变化是由于尺寸减小后大量的表面及各种缺陷出现,使体系能量升高,从而引发局域应变和能量致密化所造成的[17]。因而纳米化后HfO2的结构呈现高应力的一个状态。在升温过程中,减小的Hf-O-Hf键角逐渐被原子热振动的加剧而弛豫释放,所以该Hf-O-Hf键角出现了与体相相比显著增加的趋势。因此结果出现了纳米化后a、c轴方向上热膨胀增大,b轴方向上热膨胀性减小的现象。
3 结 论
本文采用溶剂热法制备了4 nm,尺寸分布均一的HfO2纳米颗粒,X射线同步辐射粉末衍射显示纳米HfO2具有与体相一致的单斜相结构,由于尺寸减小的原因出现衍射峰宽化。通过X射线原子对分布函数拟合提取出纳米和体相HfO2的晶体结构和热膨胀数据。HfO2的热膨胀性显示较强的各向异性。与体相材料比较,纳米HfO2沿a、c轴的热膨胀性增大,b轴热膨胀性稍微减小,总的结果是体积的热膨胀性增大。进一步结构分析表明尺寸效应使纳米HfO2晶体结构中Hf-O-Hf次近邻键角减小,导致单胞沿a、c轴方向上被压缩,b轴方向上被拉伸。升温过程中该高应力状态逐渐被原子热振动的加剧而弛豫释放因而呈现出与体相比增加的趋势。该项工作为改善纳米HfO2器件的耐热冲击性能等相关物性提供了重要的结构支撑。
猜你喜欢
中学化学(2024年3期)2024-06-30 15:19:19
中学生数理化·八年级物理人教版(2023年10期)2023-11-30 01:57:54
机电安全(2022年5期)2022-12-13 09:22:26
高中数理化(2022年16期)2022-09-14 13:57:06
科学(2020年1期)2020-01-06 12:21:34
数学物理学报(2019年5期)2019-11-29 07:46:50
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:38
数学物理学报(2017年5期)2017-11-23 07:51:09
潍坊学院学报(2016年6期)2016-04-18 13:56:55
