
补肾强身片质量标准提高研究
2019-10-23 14:24杨琪琪朱旭江王兰霞
中国民族民间医药·上半月 2019年7期
关键词:含量测定
杨琪琪 朱旭江 王兰霞

【摘 要】 目的:提高补肾强身片质量标准。方法:对淫羊藿TLC鉴别方法进行改进,新增金樱子、女贞子、菟丝子TLC鉴别;用替代对照品法测定方中原儿茶酸、绿原酸、金丝桃苷、朝藿定C、淫羊藿苷5种指标成分的含量;结果:TLC斑点清楚,阴性无影响;各指标成分在相应范围内线性关系良好;平均回收率分别为:原儿茶酸94.59%,绿原酸92.82%,金丝桃苷96.41%,朝藿定C 96.37%,淫羊藿苷96.59%(RSD<3.0%)。结论:该研究方法可为其质量控制提供参考。
【关键词】 补肾强身片;薄层鉴别;替代对照品法;含量测定
【中图分类号】R282.1 【文献标志码】 A【文章编号】1007-8517(2019)13-0028-06
補肾强身片具补肾强身之功,君药淫羊藿补肾壮阳,臣药狗脊补肝肾、强腰膝,菟丝子补阳益阴、固精缩尿、明目止泻,佐药女贞子滋肾明目,金樱子固精止遗[1-2]。原标准中有显微鉴别,化学反应及淫羊藿TLC鉴别,未见含量检测;相关文献[3-7]多数只有淫羊藿TLC鉴别和单一成分的含量测定;个别文献[8-10]对该成药中的化学成分做了定性分析,并对其中6种化学成分进行了定量分析。经验证,原标准中淫羊藿TLC鉴别专属性差,不能很好地重现,故改进淫羊藿鉴别方法,同时新增菟丝子、金樱子、女贞子的TLC鉴别,使其定性更准确。其次,本研究选淫羊藿苷为替代对照品[11-12]同时测定处方中5个指标成分的含量,以期在多组分含量测定过程中减少对照品的使用,为补肾强身片标准的制定提供更强有力的依据。
1 仪器与材料
1.1 仪器 高效液相色谱仪:Waters 2998(美国Waters);不同色谱柱:Agilent 5 TC、SunFire TM、Capcell pak均柱为C18(250mm×4.6mm,5μm)柱;恒温水浴锅(常州国华电器,HH-6);十万分之一电子天平(MS205DU)、万分之一电子天平(ME204)均为瑞士梅特勒公司;超声波清洗器(昆山市超声波仪器,KQ-500DE);薄层色谱成像仪(瑞士CAMAG,Visualizer 2)。
1.2 材料 对照药材:淫羊藿(121032-200501)、菟丝子(121232-201102)、女贞子(121041-200703)、金樱子(121047-200302)及对照品:淫羊藿苷(110737-201516,94.2%)、金丝桃苷(111521-201205,93.3%)、原儿茶酸(110809-200604)、绿原酸(110753-201716,99.3%)、朝藿定C(111780-201804,92.6%)均购于中国食品药品检定研究院;硅胶G板(青岛海洋化工);磷酸(分析纯);实验水(超纯水);甲醇、乙腈(进口色谱纯,99.9%)。补肾强身片(批号:1301、2301、3301、4301、5301、6301)及各药材阴性样品均由康县独一味生物制药有限公司提供。
2 方法和结果
2.1 薄层鉴别
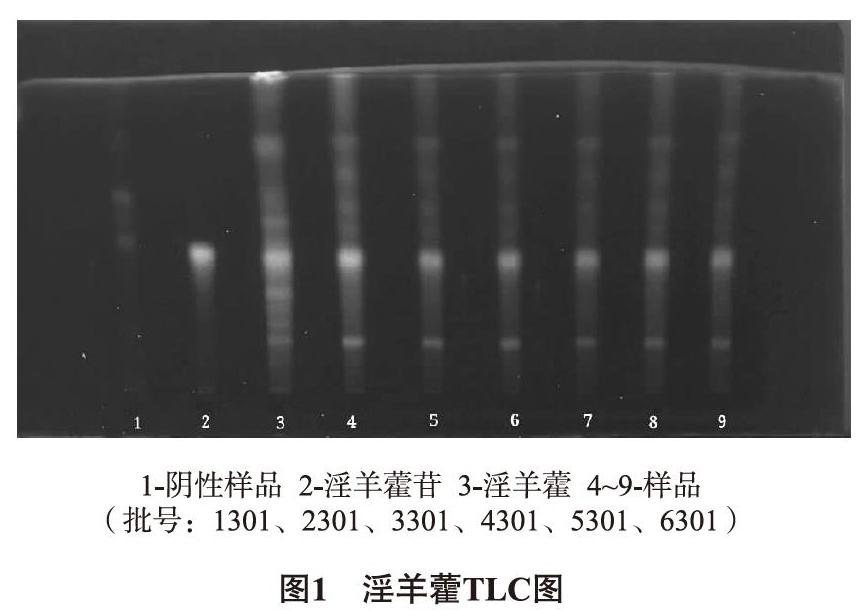
2.1.1 淫羊藿TLC鉴别 ①样品溶液的制备:各批次样品约5g,研细,25mL乙醇水浴锅回流半小时,放至常温后过滤,蒸干滤液,加水35mL少量多次溶解残渣,先用乙醚萃取两次(20mL/次)去除色素,水层再用乙酸乙酯萃取两次(25mL/次),弃去水层,蒸干,2mL乙醇溶解残渣,即得。②对照药材溶液的制备:取1g淫羊藿,同①项下处理,即得;另取适量淫羊藿苷,制成1mg/mL的对照品溶液。③阴性样品溶液的制备:取适量淫羊藿阴性粉末,同①项下处理,即得。吸取淫羊藿药材5μL,各批次样品1μL,淫羊藿苷3μL,阴性样品1μL,点于G板上,用乙酸乙酯-丁酮-甲酸-水(10∶2∶1∶1)展开[13],晾干后喷1%AlCl3试液,105℃烘干,紫外光下观看。TLC图显示,在和对照同一位置可见黄色斑点,阴性样品没有影响。如图1所示。
2.1.2 菟丝子的TLC鉴别 ①样品溶液制备:各批次样品约5g,研细,置于三角瓶中,加40mL甲醇回流半小时,放至常温后过滤,蒸干,加水35mL少量多次将残渣全部溶解,三氯甲烷萃取2次(20mL/次),三氯甲烷液蒸干,用1mL三氯甲烷-无水乙醇(2∶3)混合液溶解残渣,即得。②对照药材溶液制备:菟丝子1g, 30mL甲醇回流半小时,放至常温后过滤,滤液浓缩至1mL,即得。③阴性样品溶液的制备:取适量菟丝子阴性粉末,同①项下处理,即得。吸取菟丝子药材4μL,阴性溶液2μL,各样品溶液4μL,点于G板上,用三氯甲烷-甲醇(20∶0.5)展开,晾干后喷磷钼酸试液,110℃加热显色。TLC图中,在菟丝子对照的对应位置可看到同样的绿色斑点(白光),阴性样品没有影响。如图2所示。
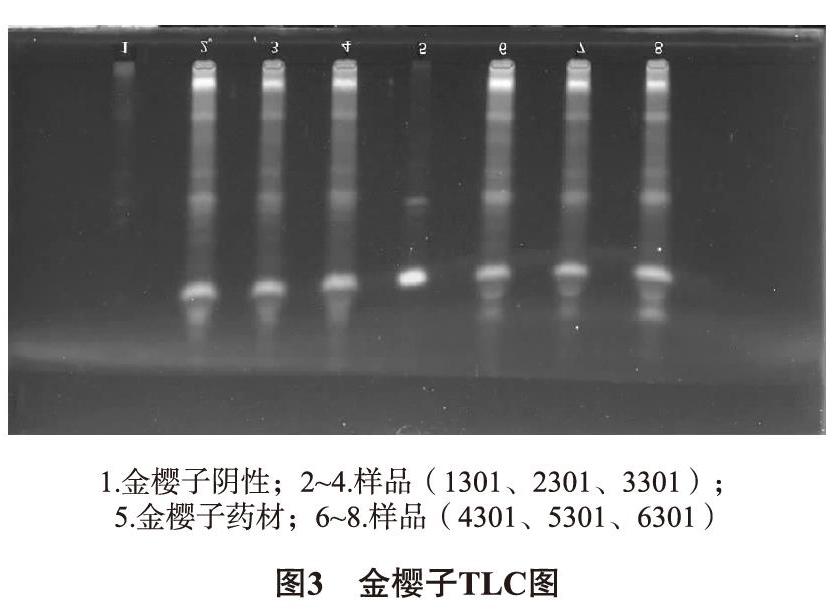
2.1.3 金樱子的TLC鉴别 ①样品溶液制备:各批次样品约5g,研细,置于三角瓶中,40mL乙醇超声半小时,滤过,蒸干,加水30mL少量多次溶解残渣,乙酸乙酯萃取两次(25mL/次),弃去水液,蒸干, 2mL甲醇溶解残渣,即得[13]。②对照药材溶液制备:取1g金樱子,同①方式提取,即得。③阴性样品溶液的制备:取适量金樱子阴性粉末,同①项下处理,即得。吸取阴性溶液1μL,金樱子药材1μL,各样品溶液6μL,点于硅胶G板上,用三氯甲烷-乙酸乙酯-甲醇-甲酸(10∶0.2∶0.2∶0.5)展开,紫外光下观察。TLC图中,在金樱子对照的对应位置可看到同样斑点,阴性样品无影响。如图3所示。
2.1.4 女贞子的TLC鉴别 ①样品溶液:取金樱子样品溶液。②对照药材溶液制备:取0.5g女贞子,30mL三氯甲烷超声半小时,滤过,三氯甲烷液蒸干,2mL甲醇溶解残渣,即得[13]。③阴性样品溶液的制备:取适量女贞子阴性粉末,同“2.1.3”项下①处理,即得。吸取阴性溶液1μL,各样品溶液6μL,女贞子药材6μL,点于G板上,用三氯甲烷-乙酸乙酯-甲醇-甲酸(5∶5∶1∶1)展开,喷10%硫酸乙醇,110℃加热显色。TLC图中,在女贞子对照的对应位置可看到同样的红褐色斑点,阴性样品没有影响。如图4所示。
2.2 含量测定
2.2.1 色谱条件 流动相:乙腈(B)-0.5%磷酸(C);梯度洗脱:0~8min,6% B;8~15min,6%~11% B;15~23min,11%~18% B;23~27min,18%~21% B;27~33min,21%~27% B;33~47min,27% B;47~48min,27%~6% B;48~60min,6% B;柱温:30℃;流速:1.0mL/min;进样量:10μL;波长:270nm。色谱图如图5所示。
2.2.2 对照溶液配制 用称量舟分别精密称取原儿茶酸16.30mg, 21.51mg,金丝桃苷18.96mg,朝藿定C 20.31mg,淫羊藿苷20.27mg分别用甲醇定容至50mL。另精密吸取原儿茶酸1mL、绿原酸1mL、金丝桃苷1mL、朝藿定C 5mL、淫羊藿苷4mL于25mL量瓶中,用甲醇定容至刻度,得浓度分别为0.01304mg/mL、0.017087544mg/mL、0.014151744mg/mL、0.07522824mg/mL、0.061101888mg/mL的混合对照品溶液。
2.2.3 样品溶液的制备 各批次样品精密称量1.0g,置于三角瓶中,每批两份平行样,精密加25mL稀乙醇,称重,超声1h,冷却后补足失重,混合均匀后静置,取续滤液。
2.2.4 专属性 取混合对照溶液10μL、样品溶液10μL,按上述色谱条件进样测定,结果显示,在相应保留时间,样品和对照品相应位置检出色谱峰。如图5所示。
2.2.5 线性关系 精密吸取5、7、9、10、11、13、15μL混合对照品溶液进样测定。纵坐标(Y)为峰面积,横坐标(X)为进样量,得线性关系,见表1;各指标成分线性关系图,如图6~10所示。
2.2.6 精密度 精密吸取10μL混合对照品溶液,连进6针,测得各成分平均峰面积:原儿茶酸298642(RSD=0.30%)、绿原酸165236(RSD=0.30%)、金丝桃苷290493(RSD=0.30%)、朝藿定C 1499789(RSD=0.40%)、淫羊藿苷1302262(RSD=2.80%),说明精密度良好。
2.2.7 稳定性 精密吸取10μL样品(批号:3301)溶液分别在0、4、8、12h进样测定,测得各成分峰面积RSD:原儿茶酸0.05%、绿原酸1.37%、金丝桃苷1.25%、朝藿定2.20%、淫羊藿苷2.69%,说明样品在12h内稳定。
2.2.8 重复性 根据“2.2.3”方法取样品(批号:3301)制备6份,按“2.2.1”项条件进样检测峰面积,计算峰面积RSD:原儿茶酸2.25%、绿原酸0.97%、金丝桃苷1.67%、朝藿定0.17%、淫羊藿苷2.87%,说明该方法可行。
2.2.9 加样回收率 精密称取6份样品(批号:3301)各0.5g,研细,参照《中国药典》2015年版四部9101指导原则[13],根据各指标成分含量,设置高、中、低浓度对照品加入量,使其与待测成分量之比控制在1.5∶1,1∶1,0.5∶1,按“2.2.3”项处理,进样测定各指标成分峰面积并计算。结果见表2。
2.2.10 相对校正因子的计算[11] 以淫羊藿苷为内标物,计算淫羊藿苷相对于原儿茶酸、绿原酸、金丝桃苷、朝藿定C的校正因子,根据不同色谱柱测得的相对校正因子,得出综合校正因子,利用综合校正因子间接计算原儿茶酸、绿原酸、金丝桃苷、朝藿定C的含量,校正因子f值见表3。
2.2.11 含量测定 按“2.2.1”项下条件,分别测定各批次补肾强身片样品,每个批次平行测定2次,计算含量;同时采用替代对照品法计算其余4个成分含量,结果见表4。
3 讨论
淫羊藿提取过程中增加了乙醚萃取步骤,研究发现在本试验中乙醚可有效去除淫羊藿药材的色素,薄层显色时使背景颜色降低,斑点更清晰可辨。金樱子和女贞子共用同一个样品提取液,调节展开劑比例即可将二者鉴别,简化了样品提取过程中的繁琐步骤,节约了时间。进行方法耐用性考察时,用不同厂家薄层板、不同温度、不同湿度分别对其进行展开,结果表明,不同板子、温度、湿度对TLC鉴别无太大影响,本研究方法可用于补肾强身片中各药材的定性鉴别。
按含量测定项下方法:①超声:20、30、40、50、60、70和80min;②功率:100%、70%、50%;③溶剂:无水乙醇、50%乙醇、甲醇。结果功率100%+超声60min+50%乙醇峰面积最大,因此选用该条件。相关文献[9]报道,特女贞苷为女贞子活性成分,本研究方法中特女贞苷不易检出,可能是梯度洗脱未能将特女贞苷分离出来,也可能是所选的提取溶剂和提取方法未将样品中的特女贞苷提取完全,故本文未曾列出特女贞苷含量测定。本试验曾尝试梯度洗脱的同时采用切换波长的方法采集各指标成分色谱峰,以期在各指标成分波长最大吸收处测定其含量,试验发现,切换波长会使样品基线发生漂移,不利于各指标成分含量测定,由于各指标成分在270nm处均有紫外吸收,故最终选择单波长270nm为其测定波长。
试验选择淫羊藿苷为替代对照品,利用相对校正因子同时测定5种成分含量;同时用不同色谱柱对校正因子进行考察,条件允许的情况下建议使用不同的仪器、色谱柱、柱温及检测波长对其进行进一步考察,使相对校正因子更加准确可靠。
4 结论
综上所述,该试验在中药成方制剂的基础上改进了淫羊藿的薄层鉴别,同时新增菟丝子,女贞子,金樱子的薄层鉴别,确保了鉴别工作的准确性。由表4可看出,两种方法所测定的指标成分含量并无太大差别,说明替代对照品法可用于该中药制剂的含量测定,为中成药多指标含量测定在节省对照品使用方面提供了新思路。
参考文献
[1]国家卫生部药典委员会.中药成方制剂:第十五册[M].北京:人民卫生出版社,1997:99.
[2]贾波,李冀.方剂学[M].北京:中国中医药出版社,2011:109.
[3]张留芳,邱德宏.补肾强身片的鉴别[J].江苏药学与临床研究,1999,7(4):44-45.
[4]赵金玲.补肾强身片中淫羊藿苷的含量测定[J].黑龙江科技信息,2013(2):20.
[5]王志成,于远洋,崔韶婧.补肾强身片中淫羊藿苷的含量测定[J].世界最新医学信息文摘,2016,16(46):28-29.
[6]穆天慧,方慧祥,黎海存.HPLC法测定补肾强身片中原儿茶酸的含量[J].中国民族民间医药,2014,(9):13-14.
[7]方慧祥,杨坤芬,冯玉茹.RP-HPLC法测定补肾强身片中金丝桃苷的含量[J].中国药房,2015,26(3):413-415.
[8]林秀莲,宋粉云,潘玄玄,等.UPLC-Q-TOF-MS分析补肾强身片中化学成分[J].中国实验方剂学杂志,2018,24(19):61-66.
[9]林秀莲,宋粉云,李华,等.UPLC同时测定补肾强身片中6种化学成分的含量[J].中国实验方剂学杂志,2017,23(20):56-60.
[10]林秀莲,宋粉云,李华,等.补肾强身片UPLC指纹图谱[J].中成药,2017,39(3):551-555.
[11]刘云华,易进海,邵华武,等.替代对照品法同时测定川芎中丁苯酞和藁本内酯的含量[J].药物分析杂志,2012,32(5):758-762.
[12]许佳,姜涛,陈廷贵,等.替代对照品法测定连翘中连翘酯苷A和连翘苷的含量[J].药物分析杂志,2013,33(10):1792-1795.
[13]国家药典委员会.中华人民共和国药典:一部[M].北京:中国医药科技出版社,2015:46,222,327.
[14]国家药典委员会.中华人民共和国药典:四部[M].北京:中国医药科技出版社,2015:374-375.
(收稿日期:2019-04-13 编辑:程鹏飞)
猜你喜欢
考试周刊(2016年103期)2017-01-23
云南中医中药杂志(2016年11期)2016-12-26
考试周刊(2016年95期)2016-12-21
