
基质匹配溶剂校准-GC-MS/MS法测定卷烟主流烟气B[a]P
2019-10-15 11:39廖惠云王晨辉
烟草科技 2019年9期
廖惠云,王 瑞,张 华,吴 洋,曹 毅,张 媛,王晨辉
江苏中烟工业有限责任公司,南京市建邺区兴隆大街29 号 210019
苯并[a]芘(B[a]P)是行业关注的卷烟烟气7 种代表性有害成分之一[1-5]。在量化分析烟气B[a]P过程中,由于复杂烟气成分带来的干扰,加之目标物在烟气中的释放量处于纳克级的水平,特别是低焦油卷烟烟气中目标物的释放量可能更低。因此,如何有效去除卷烟烟气复杂基质的干扰,快速、准确测定其中的B[a]P 是烟气分析研究的热点[6-8]。
相较于现行国标GB/T 21130—2007[9]的方法,有不少科研工作者对其量化分析进行优化研究,比如周仕禄等[10]、史佳沁等[11]、边照阳等[12]、尉朝等[13]、王春兰等[14]、段沅杏等[15]、陈晓水等[16]、王晶等[17]、申钦鹏等[18]先后对样品前处理过程或质谱仪器进行改进完善,以期达到简化样品前处理、提高分析准确度的目的。其中为了提高操作效率,采用气相色谱-串联质谱(GC-MS/MS)方法,将样品萃取液直接进样分析成为一种技术选择,但实际验证结果表明,受基质效应影响,目标物的回收率偏低。客观说来,这些分析方法对于大批量样品的快速、准确测定仍有待完善之处。因此,在参考相关文献[19-21]的基础上,建立了采用基质匹配溶剂配制标准溶液,样品萃取溶液直接进样,使用GC-MS/MS 测定卷烟主流烟气B[a]P 释放量的方法,旨在为卷烟烟气中痕量B[a]P 的定量分析提供方法参考。
1 材料与方法
1.1 材料、试剂和仪器
18 个不同盒标焦油量的市售卷烟(其中1#~5#样品为细支卷烟,6#~18#样品为常规圆周卷烟);卷烟烟气分析用1#标准样品。
环己烷、甲醇、二氯甲烷(色谱纯,德国CNW公司);氘代苯并[a]芘(B[a]P-d12)、苯并[a]芘(标准品,≥98%,德国Dr.Ehrenstorfer 公司);500 mg/6 mL 硅胶固相萃取柱(上海安谱科学仪器有限公司)。
6890N/5975C 气相色谱-质谱联用仪、7890A/7000B 气相色谱-三重串联四极杆质谱联用仪(美国Agilent 公司);RM 20H 全自动吸烟机(德国Borgwaldt KC 公司);HY-5A 回旋振荡器(常州国华电器有限公司);KQ-500DE 超声波发生器(昆山市超声仪器有限公司);R-215 旋转蒸发仪(瑞士Büchi 公司);T201 电子天平(感量0.000 1 g,瑞士Mettler Toledo 公司)。
1.2 方法
1.2.1 标准工作溶液的配制
(1)内标溶液和标准储备液
准确称取内标物B[a]P-d12,使用环己烷为溶剂配制浓度为320 ng/mL 的内标溶液;准确称取目标物B[a]P,使用环己烷为溶剂配制浓度为200 ng/mL 的标准储备液。
(2)基质匹配溶剂
按GB/T 19609—2004[22]的要求收集20 支卷烟烟气分析用1#标准样品的烟气总粒相物,添加40 mL 环己烷,于振荡器上160 r/min 条件下萃取40 min,静置片刻,得萃取液。在对萃取液进行处理前,先对固相萃取柱进行活化,即用5 mL 甲醇进行淋洗,排干,再用10 mL 环己烷平衡,柱子筛板上保留少量环己烷。然后用活化好的固相萃取柱制备基质匹配溶剂,即准确移取10 mL 萃取液至活化好的固相萃取柱,待萃取液全部移至固相萃取柱的填料后,先用环己烷分3 次、每次10 mL 进行淋洗,再用二氯甲烷分3 次、每次10 mL 进行淋洗,弃去淋洗液;最后用10 mL 甲醇进行洗脱,收集洗脱液,视为基质匹配溶剂。
(3)标准工作溶液
分别准确移取标准储备液50、100、200、500 和1 000 μL,以及内标溶液各250 μL,用基质匹配溶剂稀释定容至10 mL 容量瓶中。该系列标准工作溶液B[a]P 浓度分别为1、2、4、10 和20 ng/mL,其中内标B[a]P-d12浓度为8 ng/mL。
1.2.2 样品处理与分析
按照GB/T 16450—2004[23]的要求收集20 支卷烟的总粒相物。将收集有20 支卷烟总粒相物的玻璃纤维滤片放入100 mL 锥形瓶中(滤片应整片放入锥形瓶内铺平,不能剪碎或撕碎),准确移入1 mL 内标溶液于滤片上,并移入40 mL 环己烷,置于振荡器上于160 r/min 条件下萃取40 min,静置片刻。将萃取液过0.45 μm 有机相滤膜,将滤液直接进行GC-MS/MS 分析。分析条件为:
色谱柱:HP-5MS(30 m×0.25 mm×0.25 μm);进样口温度:280 ℃;进样量:2 μL;进样方式:脉冲不分流进样,进样压力172.37 kPa,持续时间0.75 min;载气:氦气,恒流速度1.2 mL/min;程序升温℃(20 min);传输线温度:280 ℃;电离方式:EI 源,正离子模式;离子源温度:300 ℃;四极杆温度:均为180 ℃;碰撞气:氮气,流速1.5 mL/min,载气(氦气)流速2.25 mL/min;多反应监测(MRM)模式,详细参数见表1。
2 结果与讨论
2.1 基质匹配溶剂制备考察
前期验证实验表明,使用环己烷作配标溶剂、应用GC-MS/MS 分析B[a]P 时,受基质效应影响,烟气中目标物的回收率偏低。因此,基质效应是采用MS 法分析烟气中B[a]P 时必须考虑的问题。基质效应通常不能被消除,但一般可采用SPE净化、稀释样品提取液、采用合适的内标、标准加入法以及配制基质匹配标准溶液等[24]措施来减小其对量化分析的影响。对比现行国标方法[9]采取的措施,本着简化样品前处理过程的目的,本研究中采用配制基质匹配标准溶液的方法来减小基质效应的影响,具体思路为:使用硅胶固相萃取柱净化样品溶液时,上样液和淋洗液经接收合并、浓缩后用于MS 分析,而样品溶液中其余物质被吸附保留在固相萃取柱中,理论上被视为干扰基质。在尽可能与样品溶液中目标物存在的基质环境保持一致情况下,该部分物质经洗脱下来,再经稀释后制成基质匹配溶剂,用于配制基质匹配标准溶液。

表1 目标物的MRM 参数Tab.1 MRM parameters of B[a]P and B[a]P-d12
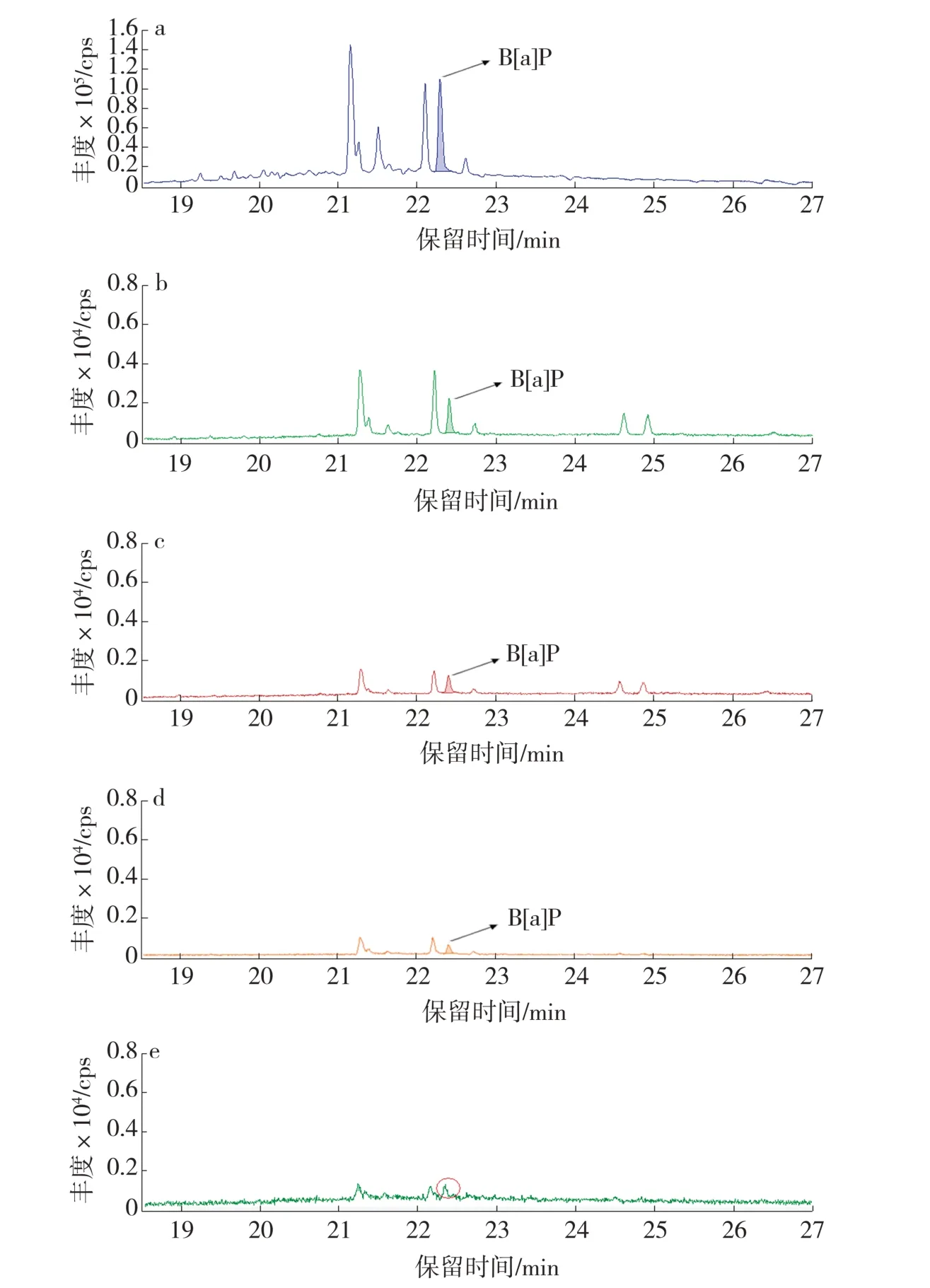
图1 不同淋洗条件下B[a]P 的提取离子对色谱图Fig.1 Ion-pairs chromatograms of B[a]P under different eluent volume conditions
为此,先行考察B[a]P 在硅胶固相萃取柱的保留行为,以期制备合适的基质匹配溶剂。具体过程为:按照1.2 节的方法,参照GB/T 21130—2007[9]的方法加入30 mL 环己烷进行淋洗,收集淋洗液并浓缩至1 mL,溶液中目标物的色谱图见图1a;再用二氯甲烷分4 次、每次10 mL 进行淋洗,分别收集淋洗液并浓缩至1 mL,溶液中目标物色谱图见图1b~图1e。色谱图1a~图1e 中B[a]P 的峰面积依次为319 715、5 114、2 488、1 486 和0(因色谱图1e中目标物的S/N 小于10,视为未检出),可见使用30 mL 环己烷和30 mL 二氯甲烷才能使目标物被完全洗脱。理论上,B[a]P 的辛醇-水分配系数lg KOW为6.04,属于非极性化合物,在硅胶固相萃取柱中是不易保留的。实际检测结果表明,用环己烷淋洗后,B[a]P 在柱子中仍有少许保留。所以在制备基质匹配溶剂时,依次用30 mL 环己烷、30 mL 二氯甲烷为溶剂淋洗萃取液,弃去淋洗液,最后直接用10 mL 甲醇洗脱,收集全部洗脱液,视为基质匹配溶剂,用于配制标准工作溶液。
2.2 仪器条件优化
2.2.1 进样方式优选
基于烟气中B[a]P 释放水平及其在萃取液中的稀释程度,为提高响应情况,在痕量分析中可加大进样量来提高灵敏度。一般来讲,溶剂在衬管内气化后有较大膨胀体积,普通压力下进样有体积限制。为此可使用脉冲模式,用以提高实际进入色谱柱的样品量,使气化后的样品更多进入色谱柱进而提高检测灵敏度;同时由于本方法中采用了脉冲进样,使得进样口压力变大,在溶剂膨胀体积和衬管体积允许条件下,将进样量由通常的1 μL 改为2 μL。在保持其他条件不变的情况下,研究进样方式对目标物响应的影响。图2a~图2d 依次为分流(分流比为10∶1)、脉冲分流(分流比为10∶1)、不分流及脉冲不分流等进样方式下目标物出峰情况。可以看出,4 种进样方式下,B[a]P 出峰情况总体较好,峰形对称,基线完全分离。相比于分流条件而言,不分流条件下目标物峰面积更大,同时信噪比也有一定程度提高。在不分流条件下,相比单纯不分流而言,脉冲不分流条件下的目标物峰面积、信噪比均显著提高。因此,考虑目标物整体出峰状况,优选脉冲不分流为色谱进样方式。
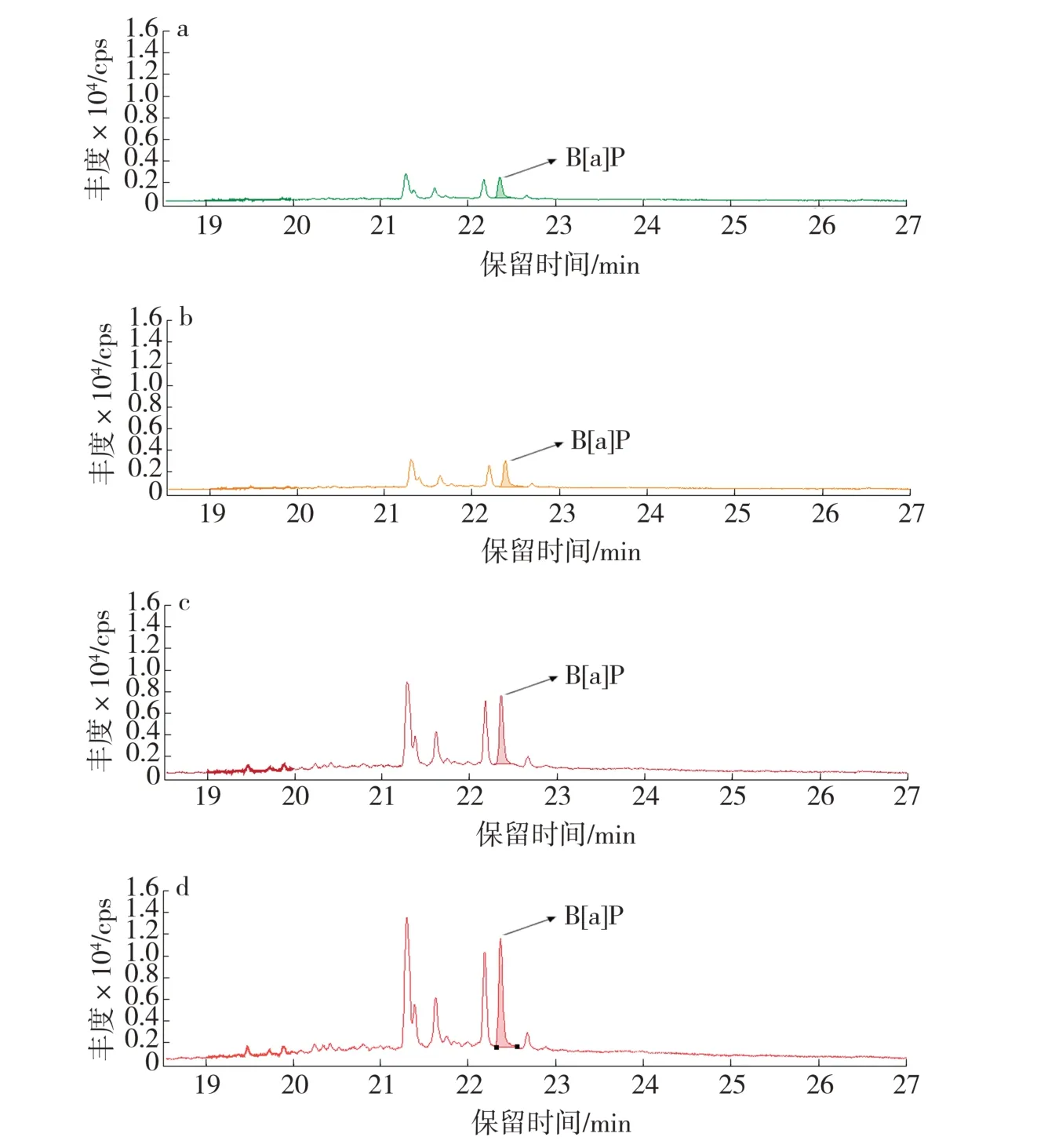
图2 进样方式对B[a]P 出峰情况的影响Fig.2 Effect of injection mode on chromatogram peak of B[a]P
2.2.2 离子对及碰撞能量的确定

图3 全扫描模式下B[a]P(a)和B[a]P-d12(b)的质谱图Fig.3 Mass spectra of B[a]P(a)and B[a]P-d12(b)under full scan mode
以高浓度标准工作溶液为对象,通过全扫描模式选择合适的母离子,B[a]P 和B[a]P-d12的质谱图如图3 所示。可以看出,B[a]P 分子离子(也就是其基峰离子)为m/z 252,B[a]P-d12分子离子(也就是其基峰离子)为m/z 264。因此,分别选择m/z 252 和m/z 264 为B[a]P 和B[a]P-d12的母离子。进一步分析,由于B[a]P 极性很小、结构非常稳定,不同碰撞能量产生的质谱图差异较大,因此在5~60 eV 碰撞能量范围条件下,采用产物离子扫描模式进行检测,以此优选产物离子及其碰撞能量。结果显示:碰撞能量小于30 eV 时,相对于母离子丰度,B[a]P 产生碎片离子的丰度均小于其10%;碰撞能量大于30 eV 时,B[a]P 母离子产生碎片离子丰度显著增大,特别是碰撞能量大于40 eV 时,母离子更大程度被打碎,产生的基峰离子为m/z 250。综合考虑各离子响应情况,确定B[a]P 的定量离子为m/z 250,辅助定性离子为m/z 226,各自优化的碰撞能量分别为50 eV 和30 eV。同理,可确定B[a]P-d12定量离子为m/z 260,辅助定性离子为m/z 236,各自优化的碰撞能量分别为50 eV 和40 eV。
2.2.3 质谱温度参数优选
考虑到最低基质配标工作溶液中B[a]P 浓度仅为1 ng/mL,当质谱仪器离子源和四极杆为常用温度时,目标物响应值低且峰形不好,对B[a]P的定量分析有一定影响。为此,以1 ng/mL 最低标样溶液为对象,探讨高离子源和四极杆温度(分别为300、180 ℃)以及常用温度条件下目标物出峰情况,具体结果见图4 和图5。可以看出,相比于常用温度质谱参数,高温质谱参数下B[a]P 和B[a]P-d12的背景信号稍微有所增加,但目标物峰形总体情况更好,峰形对称且不拖尾,信噪比提高,响应信号增强。综合考虑质谱仪器离子源温度极限值,兼顾离子源使用寿命以及定量分析结果的准确性,优选离子源和四极杆温度分别为300 ℃和180 ℃。
2.3 样品前处理优化
2.3.1 萃取液处理过程优选
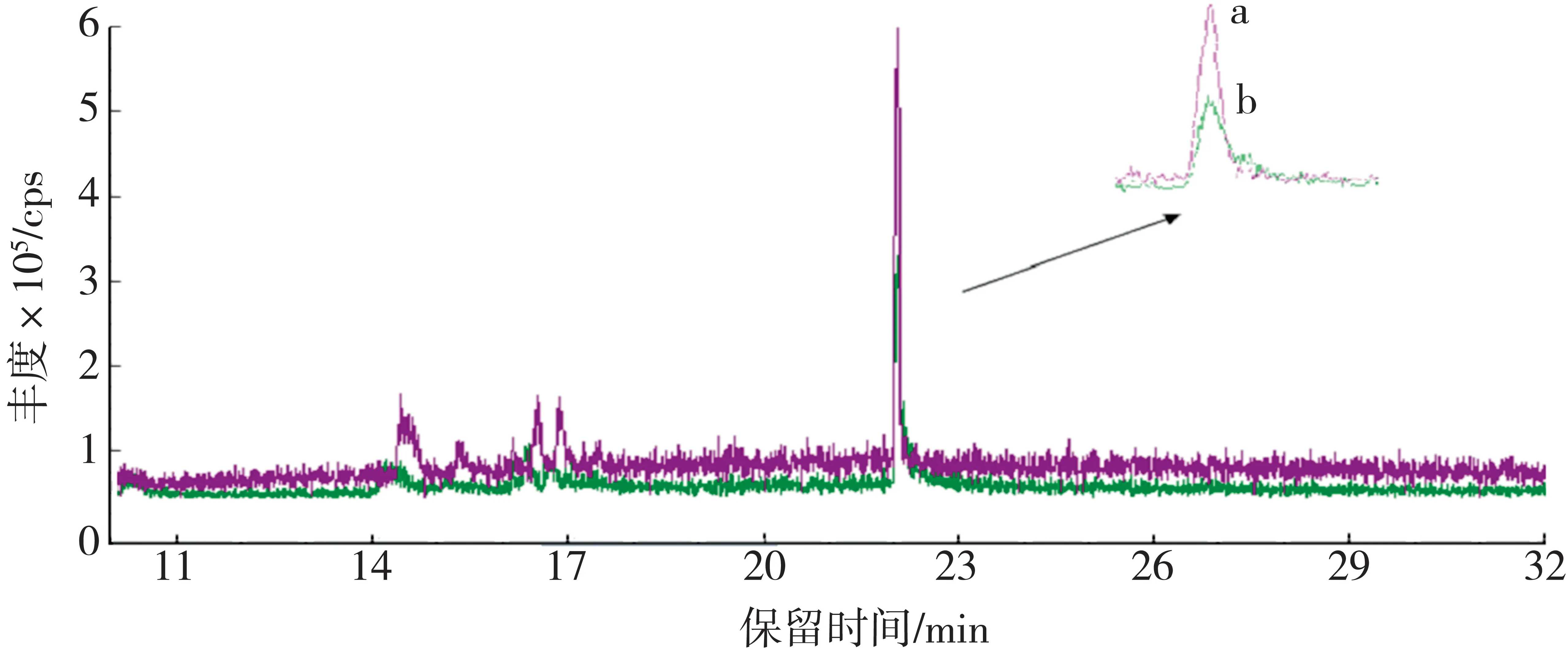
图4 不同质谱温度条件下B[a]P 的提取离子对色谱图Fig.4 Ion-pairs chromatograms of B[a]P at different mass temperatures

图5 不同质谱温度条件下B[a]P-d12的提取离子对色谱图Fig.5 Ion-pairs chromatograms of B[a]P-d12 at different mass temperatures
对于卷烟主流烟气B[a]P 的分析而言,因其释放量较低,需经过富集和分离等过程,易对检测结果带来影响。基于此,充分利用GC-MS/MS 仪器MRM 采集模式下复杂基质中目标物响应灵敏的特点,直接采用萃取溶液进行仪器分析,得到的典型样品溶液色谱图如图6a 所示。可以看出,B[a]P 峰形对称,能够与基线完全分离,基线噪声趋近于零,杂质干扰峰较少。采用现行国标方法[9]得到的色谱图(图6b),B[a]P 峰形对称,能够与基线完全分离,但是基线噪声较大,杂质干扰峰较多,信噪比相较本方法更低。由此可见,在采用GC-MS/MS 法分析时,即使直接使用萃取液进样,目标物依然能够精确定量,并且整体出峰情况优于现行国标方法。更为重要的是,样品前处理大为简便,无需对萃取液进行净化、浓缩等过程,节省了有机溶剂。
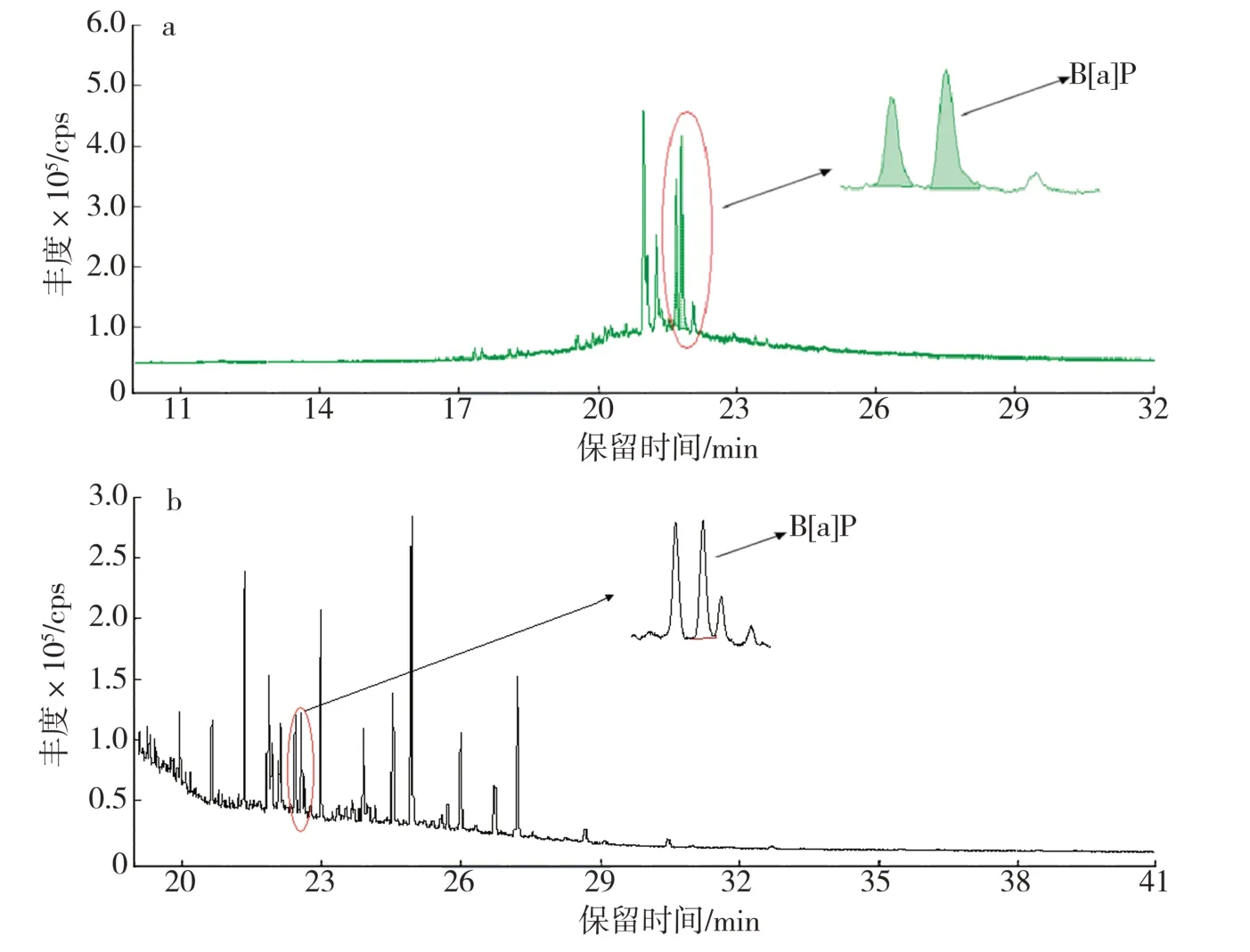
图6 不同前处理条件下B[a]P 的总离子流色谱图Fig.6 TIC chromatogram of B[a]P under different sample processing conditions
2.3.2 提取方式选择
按照现行国标方法[9]进行B[a]P 分析,采用超声萃取进行提取时,随提取时间的延长,超声波发生器内水温升高,受环己烷沸点低的影响,锥行瓶内气相压力增大,给前处理操作带来负面影响。为此,以15#卷烟样品为对象,使用GC-MS/MS 方法,将萃取溶液直接进样检测分析,比较了超声和振荡两种萃取方法对检测结果的影响,结果见表2。可以看出,随萃取时间的延长,两种萃取方式对B[a]P 检测结果影响的变化趋势较为一致,即先逐渐增大,到40~50 min 左右趋于峰值。综合考量检测结果和操作效率,选择提取方式为振荡萃取,萃取时间为40 min。

表2 萃取方式对B[a]P 检测结果的影响Tab.2 Influence of extraction method on results of B[a]P detection (ng·支-1)
2.4 方法学考察
将系列标准溶液进行GC-MS/MS 分析,求得线性回归方程及其相关参数。回归方程为Y=1.56X-0.010,决定系数R2为0.999 6,目标物在1.04~20.8 ng/mL 浓度范围内呈线性关系。以基质配标得到的最低浓度标样为对象,平行测定9 次,求其标准偏差;分别以3 倍和10 倍标准偏差对应的结果为方法的检出限(LOD)和定量限(LOQ),计算得LOD 和LOQ 分别为0.14 ng/支和0.46 ng/支,结果显示本方法比现行国标方法[9]更为灵敏。
分别以不同盒标焦油量的1#和6#卷烟样品为对象,依次进行日内精密度和日间精密度实验,计算6 次平行测定结果的相对标准偏差(RSD),结果如表3 所示。可以看出,本方法的日内RSD 介于3.13%~3.80%之间,日间RSD 介于5.41%~6.94%之间。在含有目标物的玻璃纤维滤片中分别加入低、中、高3 个水平的标准溶液,进行加标回收实验,结果见表4。可以看出,低、中、高3 个水平的加标回收率在94.62%~104.81%之间。本方法具有较好的精密度和准确度,能够满足烟气中痕量目标物的定量分析。

表3 方法的精密度Tab.3 Precision of the method

表4 方法的回收率Tab.4 Recoveries of the method
2.5 与现行国标方法的比较
分别采用本方法和现行国标方法测定了18 个不同盒标焦油量的细支和常规卷烟样品主流烟气B[a]P 释放量,结果见表5。可以看出,相比于现行国标方法,本方法检测结果的相对偏差均在±10%以内。采用SPSS 软件对两种方法的测定结果进行配对t 检验,结果表明,选取置信度为95%时,Sig.=0.216(>0.05),说明两种方法的测定结果不存在显著性差异。由此可见,对于痕量目标物的定量分析而言,两种方法的一致性较好。

表5 不同盒标主流烟气焦油释放量卷烟样品的实际检测结果Tab.5 Determination results of cigarette samples with different labeled tar levels from mainstream smoke
3 结论
①采用样品萃取溶液直接进样方式,建立了基质匹配曲线-GC-MS/MS 法快速测定卷烟主流烟气B[a]P 的方法,回收率为94.62%~104.81%,RSD 介于3.13%~6.94%之间,检出限为0.14 ng/支,定量限为0.46 ng/支。②样品对比分析表明,本方法与现行国标方法的测定结果基本一致。③本方法具有操作简便、耗费溶剂少、灵敏度高等优点,为准确、快速测定卷烟主流烟气B[a]P 释放量提供了新方法。
猜你喜欢
科学导报(2022年28期)2022-05-24
小学阅读指南·低年级版(2022年5期)2022-05-09
当代水产(2021年10期)2022-01-12
化工管理(2021年7期)2021-05-13
环境保护与循环经济(2020年4期)2020-06-08
中国特种设备安全(2019年1期)2019-03-13
发明与创新·中学生(2018年10期)2018-10-15
食品界(2017年9期)2017-09-30
中国质量与标准导报(2014年10期)2014-02-28
中国烟草学报(2012年4期)2012-04-09
