
UPLC-MS/MS测定猪肝中五氯酚残留量的不确定度评定
2019-10-15 08:36:58欧阳丽陈娅娅CN胡婷舒刘曼舒
食品与机械 2019年9期
欧阳丽- 汪 辉 何 翊 陈娅娅CN - 胡婷舒 - 刘曼舒 -
(长沙市食品药品检验所,湖南 长沙 410016)
五氯酚(pentachlorophenol,PCP)及其钠盐具有杀虫、防腐、除草等作用[1],但具有明显的生物毒性[2],美国环境保护局已将其列为114种主要环境污染物之一[3-4]。中国农业部公告第193号规定禁止五氯酚用于所有食用性动物[5]。高效液相色谱—串联质谱法因具有灵敏度高、特异性强等特点,已成为动物源性食品中五氯酚残留量检测的国标方法[6]。
作为衡量检测结果可信度的重要指标[7],测量不确定度在评定试验结果的可信性、可比性、可接受性以及检测过程中需注意的关键环节等方面都具有十分重要的意义[8]。中国实验室国家认可委员会(CNACL)对检测实验室测量不确定度评定程序的制定与应用提出了明确的要求[9]。目前尚未见对动物源性食品中五氯酚残留量的检测结果进行不确定度评定的报道。肝脏作为代谢器官,负责处理各种代谢废物与毒素,是五氯酚残留量相对较高的动物源性食品,为衡量检测结果的可信度,试验依据GB 23200.92—2016对猪肝样品进行检测,并参照现行有效的化学分析中不确定度评定方法及指南[10-12]对猪肝中五氯酚残留量的检测结果进行不确定度评定,以期提高五氯酚残留量测量结果的科学性,为五氯酚残留量的检测提供切实可行的建议。
1 材料与方法
1.1 试剂与仪器
五氯酚钠标准物质:纯度86.0%,CAS号123333-54-0,德国Dr. Ehrenstorfer公司;
甲醇、乙腈:色谱纯,德国MERCK公司;
猪肝:市场抽检样品;
实验室用水:Milli-Q超纯水;
Oasis MAX固相萃取柱:60 mg,3 mL,美国Waters公司;
超高效液相色谱—四级杆串联质谱仪:Acquity uplc®H-Class/Xevo®tq-s micro型,美国Waters公司;
快速混匀器:SK-1型,江苏金坛医疗仪器厂;
高速离心机:CT14D型,上海天美生化仪器设备工程有限公司;
固相萃取真空装置:Visiprep-DL 24型,美国Sigma-Aldrich公司;
超声波清洗器:AS3120型,天津奥特赛恩斯仪器有限公司;
全自动高通量平行浓缩仪:MV5型,美国Labtech公司;
分析天平:XSE205型,德国Mettler-Toledo公司。
1.2 方法
1.2.1 五氯酚标准储备液的制备 精确称取五氯酚钠12.59 mg至50 mL棕色容量瓶中,用甲醇溶解并定容至刻度,配制成200 μg/mL的标准储备液,-18 ℃以下避光保存;用1 mL单标线吸量管移取1 mL五氯酚标准储备液于100 mL容量瓶中,用甲醇稀释并定容至刻度,配制成浓度为2 μg/mL的标准中间溶液。用1 mL单标线吸量管移取1 mL标准中间溶液于10 mL容量瓶中,用甲醇溶液稀释并定容至刻度,稀释成浓度为200 ng/mL的标准使用液。用移液器分别移取适量标准使用液于5支2 mL容量瓶中,用空白样品基质溶液稀释并定容至刻度,配制成五氯酚浓度为1,5,10,20,50 ng/mL的标准系列溶液,供超高效液相色谱—串联质谱仪测定。
1.2.2 碱性乙腈水溶液提取 称取均质试样2 g于10 mL 具螺旋盖聚丙烯离心管中,加入6 mL 5%三乙胺的乙腈—水溶液,涡旋1 min,超声提取5 min。3 000 r/min离心5 min,收集上清液于一具刻度离心管中。离心后的残渣用约6 mL 5%三乙胺的乙腈—水溶液重复上述提取步骤1次,合并上清液,混匀。
1.2.3 固相萃取净化 将所得提取溶液转入经过预处理的MAX固相萃取柱中,以1滴/s流速使样品溶液全部通过固相萃取柱,弃去流出液。依次用5 mL 5%氨水溶液、5 mL甲醇、5 mL 2%甲酸的甲醇—水溶液淋洗,弃去流出液,淋洗液全部流出后,固相萃取柱用真空泵抽干5 min。以4 mL 4%甲酸甲醇溶液洗脱,15 mL具刻度试管收集洗脱液,40 ℃水浴下吹氮浓缩至1 mL,用水定容至2 mL,混匀。溶液以0.22 μm有机膜过滤,供测定。
1.2.4 液相色谱条件 色谱柱:Acquity uplc BEH C18液相色谱柱(2.1 mm×50 mm×1.7 μm);流动相流速:0.3 mL/min;流动相:A(5 mmol/L乙酸铵溶液)+B(甲醇),梯度洗脱程序:0.00~1.00 min,40%~100% B;1.00~5.00 min,100% B;5.01~5.50 min,100% ~40% B;5.50~9.00 min,40% B;柱温:35 ℃;进样量:10 μL。
1.2.5 质谱条件 离子源:电喷雾离子源,负离子模式;毛细管电压:2.0 kV;锥孔电压:20 V;离子源温度:150 ℃;锥孔反吹气流量:60 L/h;脱溶剂气温度:350 ℃;脱溶剂气流量:700 L/h;数据采集方式:多反应监测(MRM)。仪器参数见表1。

表1 仪器参数†
† 保留时间4.12 min;a为定量离子。
1.2.6 不确定度数学模型的建立 测量不确定度数学模型:
(1)
式中:
X——样品中五氯酚残留量,μg/kg;
c——由标准工作曲线获得样品进样液中五氯酚的浓度,ng/mL;
V——样品最后定容体积,mL;
m——样品质量,g;
R——加标回收率,%;
frep——样品测量重复性;
fc标——校准标准溶液影响因子。
2 不确定度评估
2.1 各分量不确定度来源
根据检测过程与数学模型,标准溶液的配制与稀释、标准曲线拟合、样品称量、样品测量重复性、样品最后定容、样品加标回收等环节引入的不确定度为五氯酚残留量测定结果不确定度的主要来源。
2.2 不确定度分量的量化
2.2.1 标准溶液的配制与稀释引入的不确定度
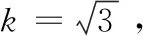
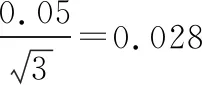
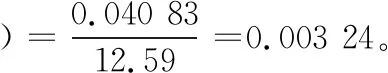

(4) 标准中间溶液配制引入的不确定度:
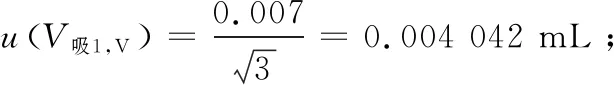
(6) 标准系列溶液配制引入的不确定度:
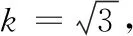
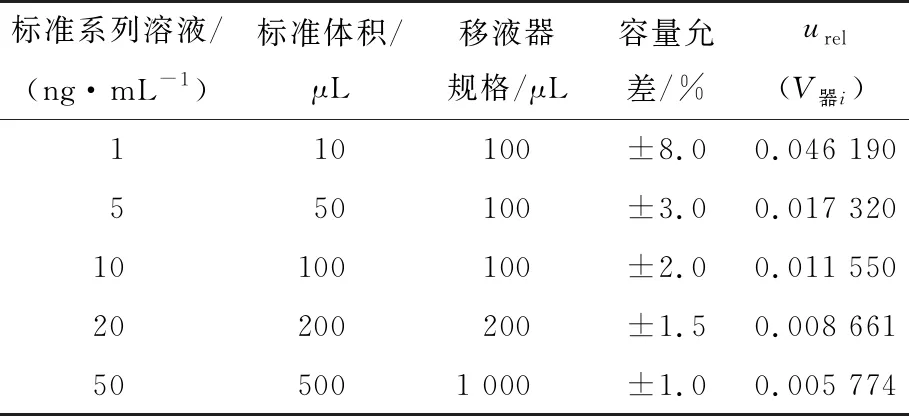
表2 移液器引入的不确定度
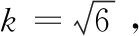
(6) 标准溶液配制和稀释引入的合成不确定度为:
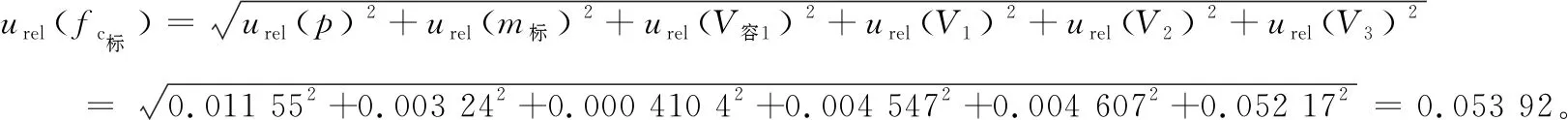
2.2.2 标准曲线拟合引入的不确定度 取标准系列溶液供超高效液相色谱—串联质谱仪测定,以浓度为横坐标,峰面积为纵坐标,获得标准工作曲线的回归方程为:A=5 355.3c+284.9(A为峰面积,c为浓度,ng/mL),相关系数为0.999 7,测定结果见表3。
根据式(2)~(4)计算最小二乘法拟合曲线引入的标准不确定度与相对不确定度。
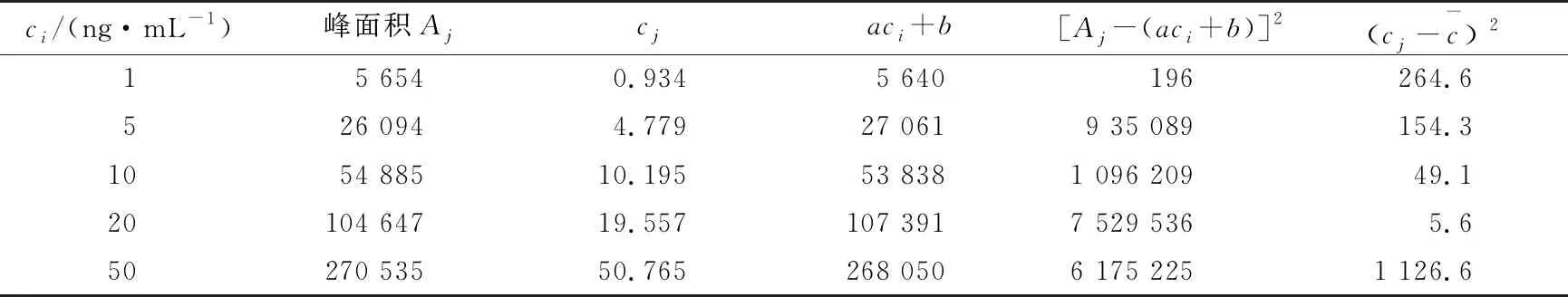
表3 五氯酚标准溶液测定结果
(2)
(3)
(4)
式中:
u(curve)——标准曲线拟合引入的标准不确定度,ng/mL;
urel(curve)——标准曲线拟合引入的相对不确定度;
S(A)——残差的标准差;
P——测定样品溶液的次数,日常检测中每个样品做2个平行即P=2;
N——获得标准曲线的标准溶液的测定次数,N=5;
α——标准曲线的斜率,α=5 355.3;
b——标准曲线的截距,b=284.9;
c0——由标准曲线拟合得到的样品溶液中五氯酚残留量的浓度,c0=14.4 ng/mL;

ci——第i个标准溶液的配制浓度,ng/mL;
cj——第j个由标准曲线拟合的标准溶液浓度,ng/mL;
Aj——标准溶液的峰面积。
将α,b,ci,Aj,N值代入式(3),得S(A)=2 290.287 8;将S(A)代入式(2),得u(curve)=0.359 1,则标准曲线拟合引入的相对不确定度μrel(curve)=0.024 94。



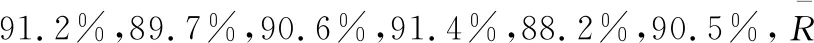
2.3 合成不确定度与扩展不确定度
将各不确定度分量进行合成得:
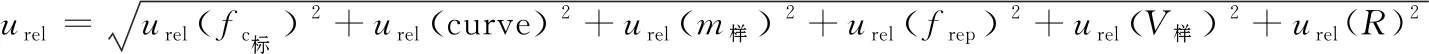
当五氯酚含量为14 μg/kg时,合成不确定度为:u=urel×c=0.099 78×14=1.4 μg/kg,选择95%置信概率,包含因子取k=2,则扩展不确定度为:U(c样)=u×k=3 μg/kg。猪肝中五氯酚残留量的测量不确定度表示为:X=(14±3) μg/kg,k=2。
2.4 主要不确定度分量的相对贡献
根据CNAS-GL006:2019规定的方法计算各不确定度分量的相对贡献,结果如表4所示。从表4可知,样品最后定容体积、样品测量重复性、标准溶液的配制与稀释引入的不确定度分量对合成不确定度的影响最大。
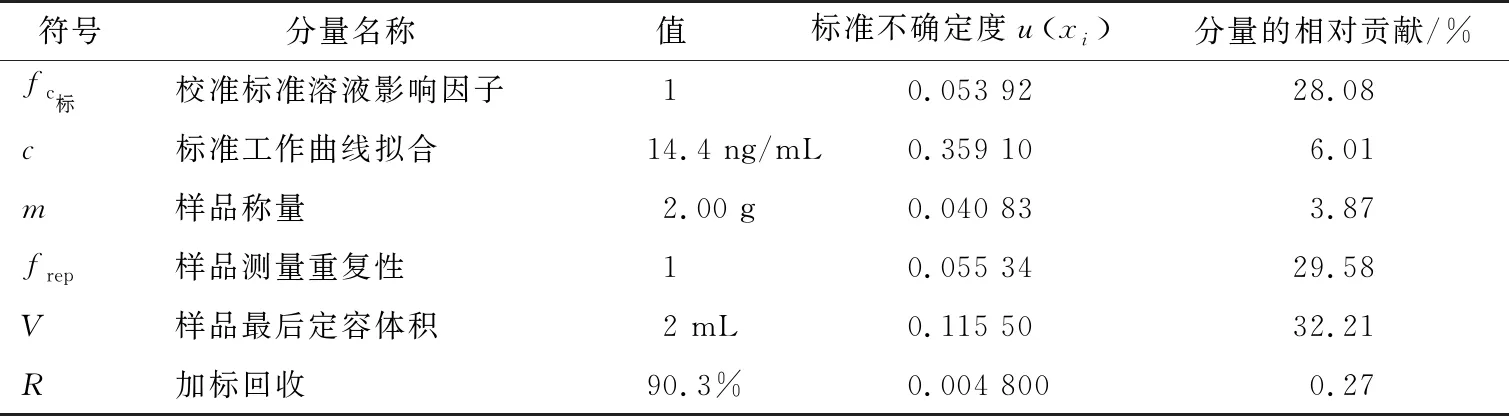
表4 各不确定度分量的相对贡献
3 结论
研究对采用UPLC-MS/MS法测定猪肝中五氯酚残留量的检测结果进行了不确定度评定。结果显示,样品最后定容体积、样品重复性测量及标准溶液的配制与稀释等人员操作环节引入的不确定度最大,此部分不确定度的相对贡献之和约为90%,为检测结果不确定度的主要来源;标准工作曲线拟合和样品称量等环节引入不确定度分量的相对贡献约为10%;加标回收引入的不确定度的相对贡献最小,仅为0.27%。因此,在检测过程中,样品制备要均匀、建议选用纯度更高的标准品、减少标准溶液的稀释步骤、定期校准仪器,减少测定结果的不确定度,以提高测定结果的准确性。
猜你喜欢
广东茶业(2019年2期)2019-06-18 10:24:24
农药科学与管理(2019年12期)2019-05-20 09:33:26
中成药(2018年1期)2018-02-02 07:20:31
价值工程(2017年31期)2018-01-17 00:34:27
电子测试(2017年12期)2017-12-18 06:35:46
水利科技与经济(2016年7期)2016-04-25 13:03:12
水利科技与经济(2016年8期)2016-04-22 03:41:22
电源技术(2015年7期)2015-08-22 08:48:52
电测与仪表(2015年7期)2015-04-09 11:40:30
中国药业(2014年24期)2014-05-26 09:00:16
