
毛竹 Phyllostachys edulis retrotransposon 7(PHRE7)转座子的克隆与鉴定
2019-09-25 02:56:04蒋政勤周明兵徐芷馨
浙江农林大学学报 2019年5期
蒋政勤,周明兵,2,郑 浩,季 航,徐芷馨
(1.浙江农林大学 省部共建亚热带森林培育国家重点实验室,浙江 杭州 311300;2.浙江农林大学 浙江省竹资源与高效利用协同中心,浙江 杭州311300)
毛竹Phyllostachys edulis为禾本科Gramineae刚竹属Phyllostachys下的一个种,在中国竹产业中有十分重要的地位。转座子是真核生物中一类重要的转座元件,在毛竹基因组中有广泛分布。转座子又可称跳跃因子,其实质是基因组染色体上能自主复制和位移的一段脱氧核糖核酸(DNA)序列,可以直接从基因组内的一个位点移到另一个位点[1],其转座的发生会导致自身碱基位置的改变和(或)拷贝数的增加。转座子根据转座方式不同可分为DNA转座子(DNA transposon)和核糖核酸(RNA)反转录转座子(retrotransposon)。DNA转座子是以DNA为中间媒介,通过 “剪切—粘贴”的方式移动,最早在玉米Zea mays中发现[2]。RNA反转录转座子是以RNA为中间媒介,依赖于反转录酶,反转录成DNA后,通过 “复制—粘贴”的方式移动,于1984年在玉米中首先被发现[3]。RNA反转录转座子又在酵母和动物基因组中被发现,后被证实在植物基因组中广泛存在[4]。例如在小麦Triticum aestivum基因组中转座子高达60%[5],玉米基因组中转座子比例高达85%[6]。RNA反转录转座子根据其结构又可以分为长末端重复序列(long terminal repeat,LTR)和非长末端重复序列(non-LTRs)[7]。 其中含有长末端重复序列的 LTR反转录转座子是目前研究较多的一类反转录转座子,几乎在所有高等植物基因组中都有分布[8],对植物基因组的结构、功能和进化有重要的作用。一个结构完整的LTR反转录转座子长度通常为2~18 kb,两端各有1个长度为100~5000 bp序列同源的长末端重复序列LTRs。LTR末端为反向重复序列,结构通常为 5′-TG-3′和 5′-CA-3′, 在 5′和 3′末端两侧通常具有 4~6 bp 的末端靶位点重复序列(target site duplications,TSDs)[9]。LTR反转录转座子除两端的反向重复的长末端重复序列外,其结构组成还包括引物结合位点(primer binding site,PBS),多嘌呤序列(polypurinetract,PPT),还有与转座机制有关的GAG(gag protein)开放阅读框和POL(polymerase)开放阅读框。目前,已在多种植物中发现具有转录活性的LTR反转录转座子,根据对活性的LTR转座子序列、进化关系和结构特征的分析,发现活性LTR反转录转座子在结构上具有相似性[10]:均含有2个 LTR区域、gag基因区、pol基因区(pr,int,rt,rh);LTR区具有1个或多个顺式调控元件;各个结构域均具有相应的关键保守氨基酸。根据转座子的长度、LTR的长度和物种分布,植物中的Ty1/Copia类转座子可分为4种类型,分别为Tork,Retrofit,Oryco和 Sire; Ty3/Gypsy类转座子可分为 6 种类型, 分别为 Tat, Athila, CRM, Reina, Del和 Galadriel[11]。LTR反转录转座子是宿主染色体结构变异和基因组进化的重要调控因子[12],与植物性状稳定表达有关。在正常的生长环境条件下,多数LTR反转录转座子不表达,呈静止状态存在,在受到逆境胁迫或DNA甲基化水平波动剧烈的情况下会被激活转座,调节植物抗逆基因的表达,从而发挥遗传可塑性来适应不同环境条件。例如,云南地方水稻Oryza sativa品种 ‘月亮谷’ ‘Yuelianggu’(传统品种,Acuche)的幼苗在高盐处理后,检测3个反转录转座子相关基因的表达水平,发现在高盐胁迫下,3个基因都被诱导快速转录,表达量均明显增加[9];陆地棉Gossypium hirsutum品系 ‘棕彩选1号’ ‘Zongcaixuan No.1’在低温胁迫下,抵御低温起关键作用的T10和T18转座子在低温胁迫后均呈上调表达[13],禾本科单子叶植物高羊茅Festuca arundinacea在干旱胁迫下,它们的甲基化程度增加,干旱诱导T10和T18转座子甲基化。DNA甲基化会抑制转座子的活性[14]。所以当植物受到非生物环境胁迫时,例如在高温、低温、高盐、辐照等逆境条件下,LTR反转录转座子会被激活并插入到目的基因中进行转座,影响周边基因的表达模式和表达量[15],帮助植物适应不同的逆境胁迫。毛竹全基因组序列分析表明:LTR反转录转座子在毛竹基因组含量最丰富,占整个基因组的37.3%(Ty1-copia,12.3%;Ty3-gypsy,24.6%)[16]。为探究毛竹LTR反转录转座子在毛竹生长受到逆境胁迫时的响应模式,本研究在毛竹基因组数据库中,选取1条具有完整结构的LTR反转录转座子,命名为PHRE7,对PHRE7转座子进行克隆与鉴定,系统分析了PHRE7在胁迫下的表达模式。本研究结果将为系统阐明LTR反转录转座子非生物胁迫下的响应机制及揭示毛竹对环境适应的分子机制奠定基础。
1 材料与方法
1.1 实验材料
植物材料:毛竹幼叶、竹笋、竹根取自浙江农林大学省部共建亚热带森林培育国家重点实验室的翠竹园,样竹长势良好。毛竹实生苗均由同一株毛竹种子培育而来。毛竹种子均取自广西省灵川同一株开花毛竹,为大小基本一致的饱满种子。
实验试剂:用于提取RNA的RNA Trizol试剂,克隆PHRE7反转录转座子的pMD18-T克隆载体,LATaq酶等聚合酶链式反应(PCR)试剂及RNA的反转录所需的Prime ScriptTMRT Master Mix和SYBRPremix ExTaqⅡ(Tli RNaseH Plus)。
1.2 实验材料处理
从同株毛竹上采集饱满且大小基本一致的优质种子,分别进行辐照、甲基化抑制剂、高温、低温、高盐等5种不同的处理,并留空白对照。
空白对照组种子处理:先用无菌水清洗种子,后用体积分数为70%乙醇浸泡消毒30 s,再用无菌水冲洗3次,然后在无菌水中浸泡24 h以恢复种子活力。取无菌培养皿,放入无菌滤纸后用无菌水润湿,再用镊子轻夹出毛竹种子,置于滤纸上静待萌发。待长至3~4片叶后(2~3周)将其转移至m(珍珠岩)∶m(蛭石)∶m(泥炭土)=1∶1∶1 的基质中生长。
辐照处理:挑选优质野生型毛竹种子450粒,150粒·组-1,分别接受 30,50和70 Gy等3个梯度的137Cs-γ射线辐照,辐照地点为浙江省农业科学院作物与核技术利用研究所辐照中心,做好不同梯度的标记,后续与不做任何处理的空白对照组在相同条件下培育到长出5片叶[17]。
甲基化抑制剂处理:毛竹种子分别用50.0,150.0和250.0 μmol·L-1的5-氮杂胞苷浸种24 h,种子数量同辐照处理一致,即每梯度150粒,但在5-氮杂胞苷处理前需用体积分数为70%乙醇消毒30 s,无菌水冲洗3次进行消毒处理。随后与空白对照组在相同条件下培育。
高温和低温处理:前面阶段与对照组的培育方法相同,待毛竹实生苗长出3~4片叶后,分别在培养箱进行高温42℃处理4 h和低温4℃处理16 h,每处理选取生长状况良好毛竹幼苗10株,后续处理与空白对照组相同。
高盐处理:前面阶段与对照组的培育方法相同,待毛竹实生苗长出3~4片叶后,分别用0.1,0.2和0.3 mol·L-1氯化钠溶液100 mL浇灌3 d[18-19],每处理选取生长状况良好毛竹幼苗10株,后续处理与空白对照组相同。
采集空白对照组及5种胁迫处理下的毛竹实生苗的叶片,每处理重复3株,用液氮速冻,于-80℃保存备用。
1.3 方法
1.3.1PHRE7反转录转座子克隆 采用十六烷基三甲基溴化铵(CTAB)法从毛竹嫩叶中提取毛竹基因组DNA[20],根据PHRE7的侧翼序列设计1对克隆引物(PHRE7-F和PHRE7-R,具体序列见表1)。以毛竹提取DNA作为克隆模板,运用以上设计的克隆引物进行PCR扩增。具体扩增反应体系设计为:LATaq酶 0.5 μL,PHRE7-F 和PHRE7-R 各 0.8 μL,2×GC 缓冲液(buffer)25.0 μL , 三磷酸碱基脱氧核苷酸混合液(dNTP mix)4.0 μL,DNA 100 ng,加无菌水补齐50.0 μL。具体反应条件设计为:预变性95℃ 5 min;变性94℃30 s,退火44℃30 s,延伸72℃5 min,35个循环;终延伸72℃10 min,4℃保存。将PCR产物在质量分数为1%琼脂糖电泳中分离,PCR产物连接到pMD20-T克隆载体上,胶回收目的片段并送生物公司测序。
1.3.2PHRE7反转录转座子编码域的相对表达量分析 分别提取毛竹未经处理叶片以及甲基化抑制剂、辐照、高温、低温、高盐等5种胁迫处理后实生苗叶片的RNA,在反转录酶的催化作用下反转录成cDNA模板,具体方法参照Trizol法[21]。在RT,INT,RH序列3个可编码蛋白结构域内分别设计特异性引物(表1),对PHRE7的表达水平进行荧光定量PCR(qRT-PCR)分析,内参基因为毛竹PheACT2[22]。荧光定量反应体系设计为 10.0 μL: 5.0 μL SYBRPremix ExTaqTMⅡ,0.4 μL cDNA,0.2 μL 底物-5(primer-5), 0.2 μL primer-3,4.2 μL无菌水[23]。反应条件如下:95℃ 7 min; 预扩增95℃ 10 s,58℃10 s,72℃15 s,共30个循环。每个样品重复3次,按照2-ΔΔt的方法计算出不同胁迫处理条件下PHRE7的相对表达量。
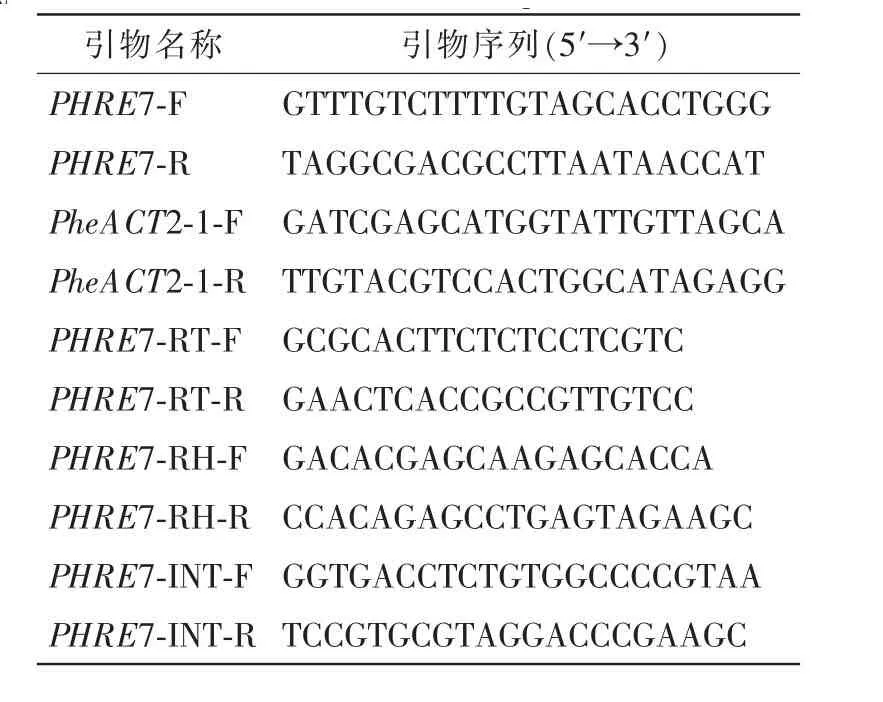
表1 引物序列Table 1 Primers sequence
1.3.3PHRE7反转录转座子结构和拷贝数分析 下载毛竹基因组数据,通过LTRharvest软件[24](http://www.zbh.uni-hamburg.de/forschung/genominformatik/software/ltrharvest.html)对LTR转座子的长度、序列等进行分析,鉴定毛竹基因组LTR反转录转座子,再利用LTRdigestion软件[25](http://www.zbh.uni-hamburg.de/forschung/genominformatik/software/ltrdigest.html),解析毛竹LTR反转录转座子结构并完成自动标注,通过cd-hit软件[26]构建序列相似的非冗余基因集,分析各序列的相似程度,确定毛竹LTR反转录转座子的拷贝数。根据毛竹LTR反转录转座子拷贝以及转座子结构,选取1个结构完整的LTR反转录转座子作为研究对象,将其命名为PHRE7。PHRE7各个结构域由LTRdigestion软件鉴定,由编码区序列翻译的氨基酸序列提交到非冗余蛋白质数据库(NCBI),通过blastp进行比对确认,并由此建立PHRE7的结构图。
1.3.4PHRE7反转录转座子插入时间分析 将PHRE7的LTR序列输入MEGA7软件[27],通过Muscle方法比对PHRE7转座子两端LTR序列的同源性,计算得分化度k。根据公式t=k/2r[28](t为插入时间,r为LTR 序列的平均替换率,1.3×10-8bp·a-1[29]),计算得PHRE7 插入时间。
1.3.5PHRE7反转录转座子LTR序列分析 通过克隆结果测序与该序列比对,确定序列完全一致性,明确开放性阅读框中的各蛋白编码区(PR,INT,RT,RH等)的位置。LTR反转录转座子的5′端LTR的U3区域都有丰富的顺式调控元件。为了分析鉴定转座子LTR区域中顺式调控元件的种类和分布情况,将LTR区域编码序列放入PlantCARE在线软件(http://bioinformatics.psb.ugent.be/webtools/plantcare/html/)[30]中,直接分析得出该区域内的各个顺式作用元件并作出标记。
1.3.6PHRE7反转录转座子进化地位分析 利用Gypsy Database 2.0(http://www.gydb.org/index.php/Main_Page)查找并下载Ty1-copia家族其他成员(Oryco,Sire,Retrofit,Tork)的代表性反转录转座子RT氨基酸序列,与PHRE7转座子的RT氨基酸序列比对,利用MEGA7软件中的Muscle方法进行同源比对[31],找出最佳模型,并通过该软件中的最大似然法(maximum likelihood,ML)构建进化树[32],分析PHRE7所在的Ty1-copia家族亚家族位置。
2 结果与分析
2.1 PHRE7的克隆
以毛竹DNA作为模板,通过PCR扩增PHRE7转座子,PCR扩增产物在质量分数为1%琼脂糖凝胶电泳后,割胶回收目的片段,送生物工程公司测序。电泳检测结果显示PHRE7转座子与预测的大小(6073 bp)基本一致(图1)。

图1 全长PHRE7转座子PCR扩增电泳图Figure 1 PCR result of full-length PHRE7
2.2 基因的序列分析
2.2.1PHRE7结构特征PHRE7转座子全长为6073 bp,测序结果与预测一致。PHRE7转座子组成为5′端和3′端的长末端重复序列和含5种酶的连续开放阅读框(ORF),结构顺序依次为5′-L-LTR-GAG-PRINT-RT-RH-LTR-R-3′(图2)。各结构长度具体如下:左端LTR(L-LTR)长度为959 bp,右端LTR(LTR-R)为3′端LTR的反向重复序列,长度也为959 bp,相似性为96.7%。计算得插入时间约为126.92万a前。开放阅读框核苷酸编码区总长度为1524 bp,共编码氨基酸507个,GAG核心区位置在1030~1744 bp,主要与反转录转座子RNA的成熟和包装有关;PR核心区为是水解酶的编码区,位置在1978~2224 bp,与反转录后多聚蛋白前体切割为功能性多肽有关;INT核心区是整合酶的编码区,位置在2365~3004 bp,用于催化反转录转座子插入宿主基因组的整个过程[33];RT核心区是反转录酶的编码区,位置在3580~4312 bp,用于催化单链RNA或DNA合成DNA的过程,是转座子转座的必不可少的酶;RH核心区位置在4624~5020 bp,用于编码核糖核酸酶H。核糖核酸酶H是水解酶的一种,负责原始RNA模板的水解。根据编码区结构顺序GAG-PR-INT-RT-RH,可确定PHRE7反转录转座子是Ty1-gypsy家族成员。通过cd-hit软件[26]对比毛竹基因组中的转座子,鉴定出PHRE7相似的序列,即其拷贝。结果显示:PHRE7存在相似的其他10个拷贝,根据拷贝位置不同,分别把10个拷贝命名(表2)为PHRE7-1,PHRE7-2,PHRE7-3,PHRE7-4,PHRE7-5,PHRE7-6,PHRE7-7,PHRE7-8,PHRE7-9和PHRE7-10,10个拷贝的结构也相对完整(图2)。

图2 毛竹基因组中PHRE7及其10个拷贝的结构分析示意图Figure 2 Structure of the PHRE7 and its copies in Phllostachys edulis genome

表2 PHRE7及其拷贝在毛竹基因组的命名及位置Table 2 Names and locations of PHRE7 and its copy in the Phyllostachys edulis genome
2.2.2PHRE7转座子LTR序列分析 在LTR序列中,顺式作用元件含量丰富。经分析得,核心启动元件有TATA-box,CAAT-box,CCAAT-box,其中TATA-box有6个,CAAT-box有21个,CCAAT-box有2个,除启动子元件外还有 2个光响应元件(4cl-CMA2b和chs-CMA2b),2个与脱落酸相关的调控元件(ABRE),3个压力响应元件(SIRE),1个参与蛋白代谢调节的顺式作用元件(O2-site)(图3)。对毛竹基因组中LTR反转录转座子PHRE7及其10个拷贝的插入时间进行分析,结果显示:PHRE7的插入时间为126.92万a前,PHRE7-1拷贝插入时间为223.08万a前,PHRE7-2拷贝插入时间为150.00万a前,PHRE7-3插入时间为142.31万a前,PHRE7-4插入时间为261.54万a前,PHRE7-5插入时间为126.92万a前,PHRE7-6插入时间为403.85万a前,PHRE7-7插入时间为442.31万a前,PHRE7-8插入时间为238.46万a前,PHRE7-9插入时间为219.23万a前,PHRE7-10插入时间为307.69万a前。经计算可知:在这11个序列中,PHRE7与PHRE7-5的插入时间相同,为126.92万a前,在11个序列中插入时间最晚,说明PHRE7是其中结构完整且最为年轻的反转录转座子(图4)。
2.2.3PHRE7转座子进化分析 为了探究PHRE7反转录转座子和植物中其他LTR反转录转座子的进化关系,利用保守性最高RT区域编码序列构建了PHRE7和其他植物典型的反转录转座子系统进化树[34]。根据进化树(图5)可知:PHRE7属于Ty1-copia家族中4个分支(Oryco,Sire,Retrofit,Tork)的Tork分支。
2.3 PHRE7在不同处理中的表达模式分析

图3 PHRE7反转录转座子LTR序列顺式元件分析Figure 3 Analysis of the cis-regulatory motifs in PHRE7
为分析PHRE7的组织表达模式,在PHRE7转座子6个结构域中选择INT,RT,RH等3个结构域进行表达量分析。PHRE7反转录转座子RT,RH,INT等3个结构域在毛竹竹根、竹叶、竹笋等3个不同部位中均有表达,且在叶中相对表达量最大,在竹笋和根相对表达丰度依次降低(图6)。
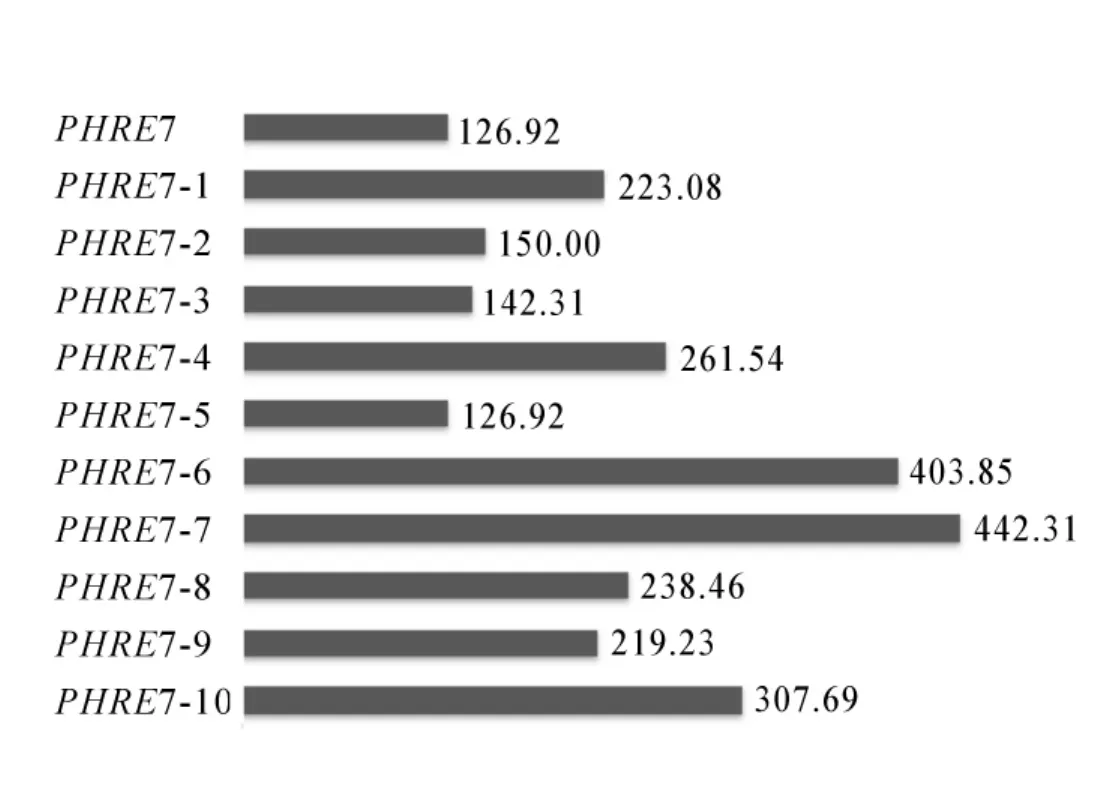
图4 PHRE7反转录转座子及各个拷贝的插入时间Figure 4 Insertion time of PHRE7 and its copies

图5 PHRE7反转录转座子系统发育树Figure 5 Phylogenetic tree of PHRE7 and other plant retrotransposons

图6 PHRE7反转录转座子INT,RT, RH等3个结构域在毛竹竹笋、竹根、竹叶中的表达量Figure 6 Expression levels of INT,RT and RH domains of PHRE7 in root,shoot and leaf of Phyllostachys edulis
PHRE73个结构域(INT,RT,RH)在竹根、竹叶、竹笋不同部位的表达量上有差异,竹叶中的表达量均为最高。为了探讨不同非生物环境胁迫条件对PHRE7反转录转座子表达的影响,研究通过定量荧光PCR对经过不同处理(辐照、甲基化抑制剂、高/低温、高盐)的毛竹实生苗叶片中PHRE7转座子的3个结构域(INT,RT,RH)表达进行了定量检测和分析。
在0.1,0.2,0.3 mol·L-1高盐处理后,高盐胁迫下PHRE7的RT,RH和INT等3个结构域表达量呈现先上升再下降的趋势,在0.1~0.2 mol·L-1时呈上升趋势, 0.2 mol·L-1左右各结构域达到表达量最高值,0.2~0.3 mol·L-1表达量逐渐下降。3个结构域中INT结构域表达量变化幅度较大,RT和RH结构域表达量相对较接近,且变化幅度相对较小(图7A)。
经 50.0, 100.0, 150.0 μmol·L-1浓度甲基化抑制剂处理, 各结构域的相对表达量随甲基化抑制剂浓度的增大逐渐升高。50.0 μmol·L-1处理组相对空白对照组表达量显著提升, 100.0 μmol·L-1与 150.0 μmol·L-1分别与前一甲基化抑制剂浓度处理组相比,表达量也均有上升。3个结构域中INT表达量最高,RT结构域表达量升高幅度比 INT小(图7B)。
不同强度的辐照胁迫下,INT,RT,RH等3个结构域表达量均高于对照组。在30 Gy辐照强度下,各结构域表达量达最大值,后随着辐照强度的增加,各结构域表达量呈下降趋势。INT结构域中30 Gy辐照处理后的表达量与50 Gy辐照处理后的表达量比值接近于2,RH和RT结构域在50 Gy或70 Gy辐照强度处理下表达量均相近,INT结构域相对表达量与RH和RT结构域相比下降幅度最大(图7C)。
4℃低温和42℃高温胁迫处理后,PHRE7反转录转座子的INT,RT,RH等3个结构域的表达量均有提高,且高温处理对表达量影响更大,上升更为显著,表达量约为低温处理的1.5倍。RT和INT结构域表达量上升幅度高于RH(图7D)。
3 讨论
PHRE7是1条结构完整的反转录转座子,按照结构域排列顺序鉴定属于Ty1-copia分支,具备转座所需的所有酶和相似度极高的长末端重复序列(96.7%)。PHRE7的LTR中含有丰富的顺式作用元件,在外界的非生物环境的胁迫下可能会诱导转座子激活进行转座。PHRE7反转录转座子的插入时间为126.92万a前,在植物基因中,很多五六百万年前插入的转座子仍存在活性,例如水稻愈伤组织中的Tos17反转录转座子插入时间约在700万a前,现仍能够进行转座[35]。相比而言PHRE7较为年轻,故推测其是仍具有转录活性的反转录转座子。

图7 不同处理条件下PHRE7反转录转座子INT,RT,RH等3个结构域的表达量Figurte 7 Expression levels of the INT,RT and RH domains of PHRE7 under different treatments
PHRE7反转录转座子各结构域完整,该转座子其他拷贝存在不同结构域间的缺失。转座致使宿主基因组出现断裂和重排的现象,还会影响宿主基因表达,导致宿主基因组处于不稳定的状态。以上现象会激活宿主对LTR转座子活性抑制机制,导致LTR转座子在进化发展过程中,各结构域间出现基因片段丢失现象,控制该转座子的自主转座。所以,PHRE7反转录转座子及其10条拷贝可能是由共同祖先演化变异而来,而PHRE7反转录转座子在宿主基因调节机制下依然保持其完整的结构。
PHRE7在毛竹各部位的表达存在差异性。在毛竹竹笋、竹根和竹叶等3个不同部位表达量对比中,PHRE7在竹叶中的表达量相对较高。有研究证明:活性LTR反转录转座子在不同部位间的表达具有选择性和特异性,可能与LTR反转录转座子长末端重复序列中的顺式作用元件和其他元件的存在有关,各类元件与调控物质的作用不同,对LTR转座子的表达量也存在一定影响。
毛竹实生苗在甲基化抑制剂、辐照、高盐、高温和低温等5个不同处理后,3个结构域表达量均比对照高,表明PHRE7是1个具有转录活性的转座子,转录活性受外界环境变化的影响。
LTR反转录转座子的转座活性与其甲基化程度有较大程度的关联。通常情况下DNA甲基化可以作为基因沉默的一种表观遗传标记[36]。在毛竹基因组中,编码区占比较小,非编码DNA序列[37]大量存在。为保证基因表达的准确性,植物体要尽可能减少非编码区基因对转录过程的干扰,因此在正常条件下,非编码区DNA较少或基本不表达。而在甲基化抑制剂作用下,许多存在于非编码区的转座子会通过去甲基化作用解除限制使非编码DNA得以激活表达[38],因此发生甲基化抑制剂作用下各结构域表达量显著上调现象,且甲基化抑制剂的浓度越高,表达量越高,呈正相关。
逆境胁迫致使毛竹体内的激素水平、离子水平和甲基化水平等都发生一定程度的变化,从而影响转座子的转座活性[39],引起表达量的变化,这也是LTR反转录转座子对逆境做出的一种适应[40]。辐射能引起植物变异。本研究结果显示:辐照条件下,PHRE7转座子表达量比空白对照组要高,但是随辐照强度的增强表达量发生下调,其可能原因是高辐射对毛竹实生苗造成不可恢复的伤害,致使毛竹的自身调节功能不足以抵御外界辐照的伤害,各结构域表达量随辐照强度的增强而下降。
在高盐胁迫下,植物体内含水量会下降,水分过低会引起植物代谢和光合作用异常[41]。PHRE7的LTR序列中存在光响应元件(4cl-CMA2b和chs-CMA2b),表明PHRE7转座子转录可能受光合作用调节。在高盐作用下,PHRE7表达量随盐浓度的升高呈现先上升后下降趋势。其可能原因是盐浓度提升致使毛竹实生苗光合作用异常[42],转座子得以激活表达,PHRE7结构域表达量升高。
PHRE7在4℃低温和42℃高温处理下,表达量较对照组均有上调,且42℃较4℃更为显著。温度胁迫是植物中常发生的胁迫类型,温度对催化各类植物生理活动的酶活性有显著影响,但极端温度胁迫会造成基因组甲基化水平下降。在此效应下,PHRE7结构域的表达量呈上调变化。
本研究对毛竹反转录转座子PHRE7的序列、结构以及在典型非生物环境胁迫下的表达量进行了系统的研究,发现PHRE7的表达量会在环境影响下有所变化,能对外界环境胁迫做出响应,是具有转录活性的LTR反转录转座子,但具体的响应机制尚未清楚,仍需进一步的研究。
猜你喜欢
林业科学(2022年1期)2022-03-23 06:56:24
中国蜂业(2021年5期)2021-05-22 02:59:26
意林·少年版(2020年13期)2020-08-02 11:02:50
中国生殖健康(2018年1期)2018-11-06 07:14:38
浙江农林大学学报(2016年6期)2016-12-12 12:01:32
福建农业科技(2015年1期)2015-02-27 10:20:39
福建农业科技(2015年1期)2015-02-27 10:20:38
华东理工大学学报(自然科学版)(2014年5期)2014-02-27 13:49:27
计算机应用文摘(2011年8期)2011-04-29 00:44:03
计算机应用文摘(2010年30期)2010-04-29 00:44:03
