
硫酸氢酯类药剂浮选性能的量子化学研究
2019-09-20 09:33慕红梅马海涛
价值工程 2019年23期
慕红梅 马海涛


摘要:本文运用密度泛函理论,选取B3LYP/6-311+G(d,p)方法基组,对碳原子数在15~21的硫酸氢酯类浮选药剂进行了计算研究。通过几何参数优化、电荷分布的变化,推测碳原子数在15~21的硫酸酯类浮选药剂对金属离子浮选性能的好坏,这为设计浮选药剂提供了较好地途径。
Abstract: In this paper, the density functional theory is used to select the B3LYP/6-311+G(d,p) method base group to calculate the hydrogen sulphate flotation reagent with 15~21 carbon atoms. Through the optimization of geometric parameters and the change of charge distribution, it is speculated that the flotation of a sulfate-based flotation agent with a carbon number of 15-21 is good for the metal ion flotation performance, which provides a better way to design flotation reagents.
关键词:量子化學;密度泛函理论;浮选药剂
Key words: quantum chemistry;density functional theory;flotation reagent
中图分类号:TD923 文献标识码:A 文章编号:1006-4311(2019)23-0272-02
0 引言
矿产资源是人类赖以发展的物质基础。我国虽矿产资源丰富,但随着开发规模扩大,可利用矿物形势越来越严峻,而经济发展对矿物材料仍有较大需求[1],所以我们现在面临着经济发展与矿产资源紧缺的新局面。泡沫浮选法[2]是目前重要的浮选方法,其中捕收剂的品种和质量直接关系到浮选工艺效果的好坏[3]。所以,根据特定的浮选任务,设计出新型高效、廉价环保的浮选药剂,对我国矿物的综合回收利用以及生态环境安全具有非凡意义。开发新型浮选药剂的方法有物理化学方法、拓扑学方法和分子模拟方法[4]。近年来发展比较迅速的分子模拟方法是量子化学方法和分子动力学方法,采用分子模拟技术能降低浮选新药剂开发的经济成本,提高设计的效率和质量,对现存的复杂难选矿物的开发利用具有重要的理论和实际应用价值。
近年来,国内外基于密度泛函理论的量子化学方法研究浮选药剂设计主要集中在黄铁矿和黄铜矿的分子结构、黄药、胺类等浮选药剂。
通过查阅文献,未见有关硫酸酯类浮选药剂的研究报道。本文选用15~21个碳原子的硫酸酯,研究其几何参数、电荷密度分布情况,及15~21个碳原子的硫酸酯与金属离子作用后吸附能变化,通过以上研究,希望能为浮选药剂设计提供理论依据。
1 计算方法
1.1 几何构型优化
采用密度泛函(DFT)B3LYP方法[5],6-311+G(d,p)[6]基组,对碳原子数15~21的硫酸酯进行全参数优化,得到各物质能量和电荷分布,优化后各物质与金属离子作用,得到吸附能。所有计算工作由Gaussian09程序完成[7],由Gauss View 程序从计算结构直接转换生成分子的几何构型。
1.2 硫酸酯类分子结构模型
利用Gauss View 程序分布搭建了碳原子数为15~21的硫酸酯类浮选药剂分子模型,图1和图2分别时C15分子的结构图和电荷分布图,其余碳原子数的硫酸氢酯分别在分子中增加-CH2基团就可。
2 结果与讨论
2.1 优化后能量变化
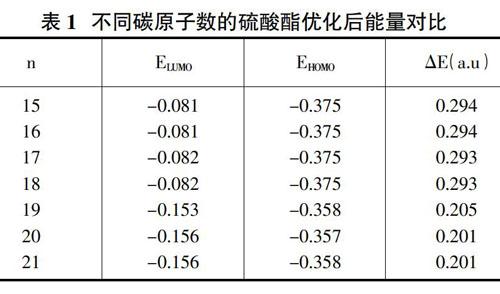
选用DFT方法计算的碳原子数为15~21的硫酸氢酯类分子的量子化学能量参数见表1,其中n为碳原子个数,ELUMO为最低未占分子轨道,EHOMO为最高占据分子轨道。
前线轨道理论指出,分子的最高占据轨道与最低空轨道决定着分子的电子得失与转移,从而决定分子间的空间取向和化学反应等性质。为了综合考虑硫酸氢酯类浮选药剂的捕收能力,可用HOMO值与LUMO值的差值即前线轨道能隙?驻ELUMO-HOMO来进行讨论,其绝对值越小,分子之间发生相互作用越容易,浮选过程中的捕收能力越强。因此,不考虑药剂与矿物的作用机理和药剂分子上所带电荷,仅从前线轨道能隙来看,碳原子数从15到21,?驻E依次降低,预计其捕收性能会越来越好。
2.2 电荷分布变化
碳原子数为15~21的硫酸氢酯中相同位置上的S、O、C原子的电荷列于表2中。图3为碳原子数为15~21的硫酸氢酯HOMO 和LUMO能量分布图。
2.3 电荷分布变化
把碳原子数为15~21的硫酸酯中相同位置上的S、O、C原子的电荷列于表2中。
从表2可看出,在不与金属离子作用前,C15~C18能量均匀分布,从C19~C21电荷开始向硫酸氢酯基转移,可推测出,随着碳原子数的增多,电子不均衡性会越明显,可能越有利于和金属离子反应。
3 结论
采用密度泛函DFT-UB3LY方法,6-311+G(d,p)基组,对碳原子数在15~21的硫酸酯类浮选药剂作用于金属离子进行了计算得出以下结论:①不考虑药剂与矿物的作用机理和药剂分子上所带电荷,但从前线轨道能隙来看,碳原子数从15到21,?驻E依次降低,预计其捕收性能会越来越好。②随着碳原子数的增多,电子不均衡性会越明显,可能越有利于吸附金属离子。
這个结论为金属选别时设计药剂提供了较大的理论支持。
参考文献:
[1]王淀佐,姚华军,贾文龙,等.有关矿产资源节约与综合利用的思考与建议[J].中国国土资源经济,2011(10):4-6.
[2]沈旭,熊崑,李柏村.浮选技术[M].重庆:重庆大学出版社, 2011.
[3]N. А Каковский,谢仁兴.浮选药剂理论和应用的进展[J]. 国外金属矿选矿,1984(04):40-47.
[4]张行荣,刘龙利,吴桂叶,等.浮选药剂分子结构设计原理概述[J].矿冶,2013,22(03):25-29.
[5]Stephens P J, Devlin F J, Chablowski C F, et al. Abinitio calculation of vibrational absorption and circular dichroism spectra using density functional force fields [J]. J. Phys.Chem.,1994,98:11623.
[6]Gronert S. The need for additional diffuse functions in calculations on small anions :the G2(DD) approach [J].Chem phys,let.,1996,252:415.
[7]Frisch M J, Trucks G W,Schegel H B, et al. Gaussian 09 ,Revision D 01.Wallingford CT : Gaussian,Inc., 2013.
