
肝细胞癌相关转录激活因子KLFs的研究进展
2019-09-17 01:30孙建英巫梦娜姚登福
胃肠病学和肝病学杂志 2019年9期
孙建英, 巫梦娜, 姚 敏, 姚登福
1.南通大学附属医院临床医学研究中心,江苏 南通 226001; 2.南通大学医学院免疫学系
肝细胞癌(hepatocellular carcinoma,HCC)为常见恶性肿瘤之一,早期诊断难,治疗效果不理想,预后差[1]。近年研究表明,KLFs家族共有17种亚型,与前列腺癌、甲状腺癌、食管癌、胃癌、肝癌、胰腺癌、肠癌、膀胱癌、乳腺癌、宫颈癌等多种肿瘤的发生、进展关系密切[2]。在KLFs家族成员中,已发现KLF4、KLF5、KLF6、KLF8、KLF9、KLF10及KLF17 7个成员的异常表达[3],参与了HCC增殖、分化、侵袭和转移过程,作为转录激活因子通过调节基因转录调节细胞周期和细胞的增殖分化,在癌细胞分化过程中发挥着重要作用,机制复杂,但HCC相关KLFs异常,在HCC的诊断、预后及治疗等方面具有应用前景。本文主要综述了7种与HCC发生、发展密切相关KLFs的研究进展。
1 KLFs家族成员、分布、基因定位与功能
KLFs家族包含17个成员(KLF1~17,见表1)由73个氨基酸组成。KLFs家族羧基末端都被发现具有3个高度保守的锌指蛋白(C2H2)结构域,其中第一、第二个锌指结构各含25个氨基酸,第三个锌指结构含23个氨基酸,成员间高度保守,可激活和抑制靶基因转录,优先与“GC box”或“CACCC”元素位点结合[4],以细胞型和启动子依赖方式作为跨调控元件,并且含有与靶DNA结合的锌指结构域。KLFs功能各异,通过多种调控机制对靶基因进行激活或抑制,控制靶基因表达。相同的KLFs与不同的启动子结合,或参与调控的辅助因子改变,激发KLFs不同调控功能。
按照KLFs功能可分为与羧基末端结合蛋白(Ctbp)相互作用的转录抑制因子KLF3、KLF8和KLF12;具有结合脱乙酰酶能力的转录激活因子KLF1、KLF2、KLF4、KLF5、KLF6和KLF7;与共同的转录辅助抑制因子Sin3A的相互作用而产生抑制活性的因子KLF9、KLF10、KLF11、KLF13、KLF14和KLF16[5]。人体KLFs参与呼吸、免疫和血管等多系统的调节作用,此外KLFs也参与肿瘤生物活动,诱导多能干细胞,并保持多能胚胎干细胞的状态[6]。特异性蛋白(SPS)和KLFs属于转录因子家族,含有与靶DNA结合的锌指结构域。其中许多蛋白在不同的组织中表达,并具有不同的组织特异性活性和功能。SPS和KLFs不仅调节生长、发育、分化、增殖和胚胎发育等生理过程,而且调节多种疾病的发病机制,包括癌症和炎症性疾病[7]。KLF家族的成员与SPS具有结构同源性和DNA结合能力,因此常被称为SP/KLF转录因子[8],根据序列和结构相似性分为3个亚家族,亚族Ⅰ包括与Sp1最相似的9个成员分别被命名为Sp1~Sp9[9]。亚族Ⅱ包括KLF1~KLF8和KLF12,亚族Ⅲ包括KLF9~KLF11、KLF13、KLF15和KLF16。
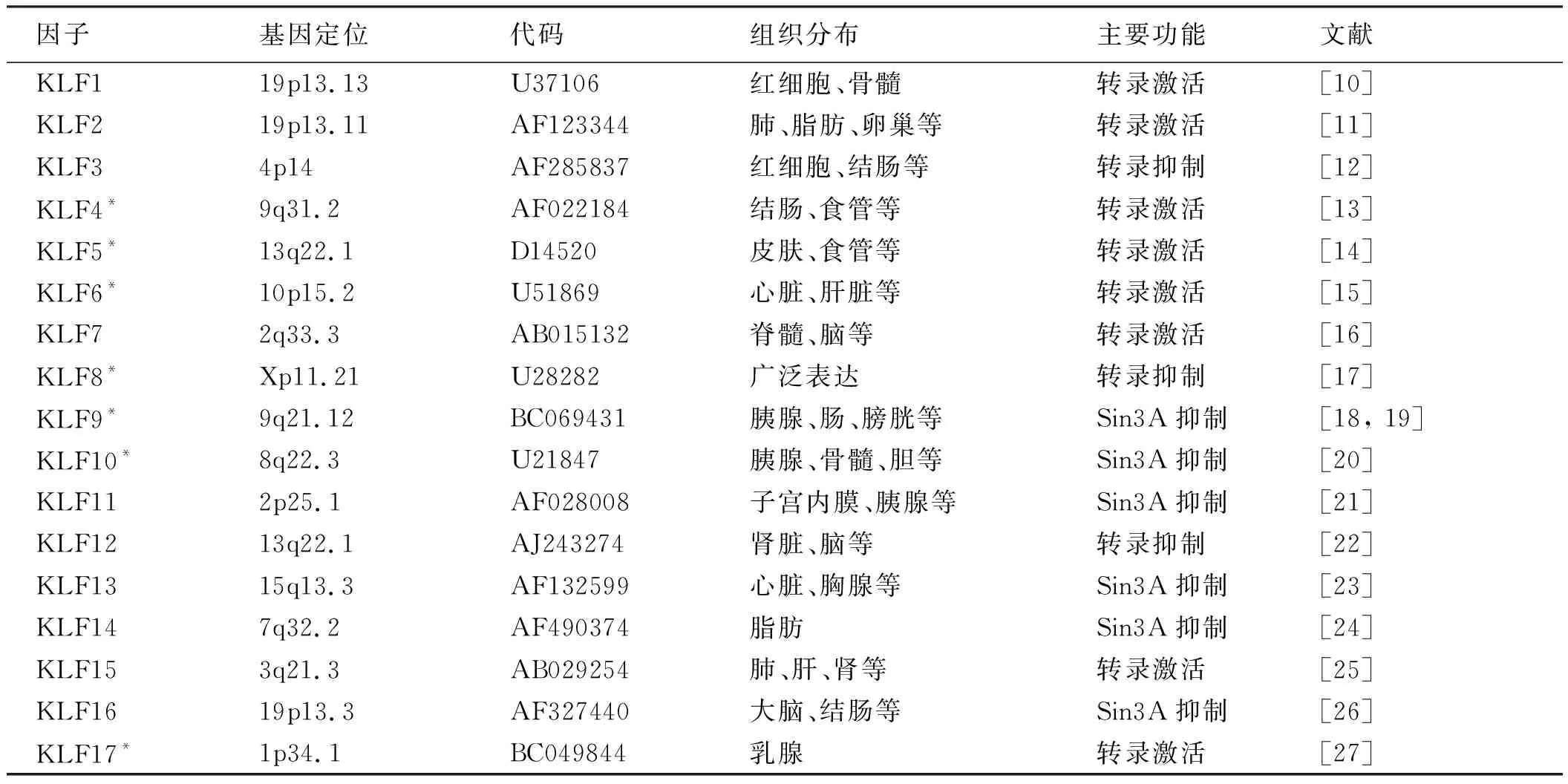
表1 KLFs家族成员的基因定位、组织分布与主要功能Tab 1 Gene positioning, tissue distribution and main function of KLFs family members
注:*:HCC相关KLFs。
2 KLFs家族成员与HCC
基础及临床研究发现KLFs在前列腺癌、甲状腺癌、食管癌、胃癌、肝癌、胰腺癌、肠癌、膀胱癌、乳腺癌、宫颈癌等多种肿瘤中发挥促癌或抑癌作用。其中KLF4、KLF5、KLF6、KLF8、KLF9、KLF10及KLF17等7个成员异常表达[3],参与了HCC增殖、分化、侵袭和转移过程,作为转录激活因子通过调节基因转录调节细胞周期和细胞的增殖分化,在癌细胞分化过程中发挥着重要作用,机制复杂。
2.1 KLF4与HCCKLF4是一种锌指转录因子,在细胞周期、增殖和凋亡中起重要作用。在HCC中,KLF4具有抑癌功能。KLF4主要在胃肠道、血管内皮细胞中表达,故称“胃肠富集型KLF”[28]。KLF4主要通过以下机制发挥抑癌基因作用:抑制Wnt/β-catenin信号通路;上调Notch通路抑制肿瘤的发生、发展;抑制肿瘤上皮间充质转化(EMT):此过程涉及多种信号通路,主要包括ERK通路活化、PI3K/AKT、Wnt/β-catenin信号通路、NF-κB通路、VEGF通路、转化生长因子-β(TGF-β)等,参与APC信号传导通路;诱导肿瘤干细胞增殖;激活p53、p21等抑癌基因[29]。
2.2 KLF5与HCCKLF5是包含3个锌指结构域和1个反式激活结构域的转录因子[30]。KLF5的DNA结合位点有一个乙酰化位点,KLF5的乙酰化状态对其转录活性很重要。KLF5可以调节多种生物过程,调控许多与细胞周期、血管生成、迁移、炎症等相关的因素,包括癌细胞的增殖、凋亡、血管生成、干性和EMT。KLF5参与几个致癌信号通路包括RAS/ERK、PI3K/AKT、p53、TGF-β的调控,并起主导作用。在调节细胞增殖和肿瘤发生中,KLF5看似对立的效果取决于细胞上下细胞信号分子间包括TGF-β的状态和可能的其他信号的活动。在肿瘤发生中,KLF5具有相反的功能,并且在一些研究中具有肿瘤抑制作用,根据细胞环境抑制和促进肿瘤生长,控制KLF5功能转换的机制仍然未知。肿瘤促进机制可作为治疗的靶点。虽然KLF5的一些转录靶点已经被识别出来,并与KLF5参与的肿瘤生长有关,但KLF5如何调控这些基因仍有待解决。
2.3 KLF6与HCCKLF6基因位于10p15,由4个外显子构成,除羧基末端3个高度保守的锌指结构域外,氨基末端还包含1个丝氨酸/苏氨酸富集及1个酸性结构域,具有转录激活功能。胎盘KLF6高表达,为癌胚抗原。KLF6基因有3个剪接变异体(SV1、SV2、SV3)[31]。在前列腺癌、神经胶质细胞瘤、结直肠癌、肝癌等恶性肿瘤中,KLF6是一种肿瘤抑制因子,不同的剪接变异体可以在不同肿瘤的起源、发展过程中发挥不同作用。KLF6 SV1在肝癌组织表达升高,为促癌基因[32],KLF6 SV2是抑癌因子[33],在肝癌组织及细胞系中发挥抗细胞增殖、促进细胞凋亡的作用。
2.4 KLF8与HCCKLF8是HCC的促癌基因。HCC浸润及转移首先是MMPs等基质金属蛋白酶降解细胞外基质,进而癌细胞通过EMT过程从原发病灶脱离,进入周围组织和血液、淋巴循环,发生浸润和转移。MMP-9是MMP家族中的一员,具有降解变性胶原和Ⅳ型胶原蛋白的功能,与HCC的进展及低生存率密切相关。研究[34]显示,KLF8蛋白表达水平与MMP-9含量呈正相关,这表明MMP-9可能是KLF8的下游靶基因,上调KLF8基因的表达导致HCC细胞株MMP-9的活性增加,促进癌细胞的浸润及转移。相关研究[17]发现,肝癌组织KLF8的表达水平与黏着斑激酶(focal adhesion kinase,FAK)表达水平呈正相关,FAK基因过表达可促进HCC的转移,表明KLF8可能受FAK调控,但具体信号通路机制尚不明确。KLF8基因在HCC中参与调控血管生成,即KLF8通过PI3K/AKT信号通路诱导VEGF的表达,KLF8高表达的HCC细胞具有更强的诱导新血管生成潜能。KLF8及β-catenin蛋白表达水平在高分化HCC中显著升高,提示KLF8参与了肿瘤浸润和转移过程中的重要信号通路Wnt/β-catenin的转录调控。一方面,Wnt3a可以刺激KLF8表达水平升高;另一方面 KLF8过表达提高了β-catenin对T-cell factor 4转录因子的招募能力,进而诱导Wnt/β-catenin信号通路靶基因c-Myc、Cyclin D1及Axin1表达。
2.5 KLF9与HCCKLF9是结直肠癌和胶质母细胞瘤中的抑癌基因,在HCC组织中,KLF9表达下调。KLF9的上调对SK-Hep1和HepG2细胞的生长有明显的抑制作用,并引起细胞凋亡[35]。KLF9通过直接结合p53启动子近端区域内的GC盒来正调节p53水平。在环己酰亚胺存在下,KLF9显著增加HCC细胞中的p53稳定性。KLF9的异位表达足以延缓肿瘤的发作并促进已建立的肿瘤在体内消退,KLF9在HCC发展中起关键作用,且KLF9的药理学或遗传活化对HCC可能具有治疗的作用。
2.6 KLF10与HCC有许多信号通路参与了肝脏肿瘤的发生。其中TGF-β/SMAD途径与KLF10在大多数情况下均被报道为抑制细胞增殖的途径之一[36]。KLF10通过产生活性氧和线粒体膜电位的丧失,介导TGF-β诱导的HCC细胞凋亡[7]。
2.7 KLF17与HCCKLF17是抑癌基因,KLF17的低表达和失活是由人类肿瘤中的微小RNA,基因突变和杂合性缺失引起的,参与肿瘤进展。KLF17低表达增加癌症转移生存力,其机制是低KLF17通过调节EMT相关基因表达参与EMT。降低的KLF17也通过上调DNA结合抑制剂1(ID1)来增加癌症转移。此外,突变型p53蛋白能够与KLF17形成复合物参与KLF17的消耗,抑制EMT基因转录并增加癌症转移。同时,KLF17下调也参与TGF-β途径的激活。60例HCC组织KLF17表达与生存率间关系,显示KLF17高表达、低表达患者5年生存率分别为63%、41%(P=0.034)。KLF17低表达组淋巴结及远处转移的概率更高。miR-9过表达促进HCC细胞转移和浸润,HepG2 HCC细胞中miR-9表达水平较正常肝细胞增高1 000倍,miR-9通过结合KLF17基因3′UTR抑制KLF17基因表达,共转染miR-9后,Hep12 HCC细胞KLF17蛋白表达下调;抑制miR-9表达后,则KLF17蛋白表达上调。KLF17直接结合波形蛋白、ZO-1、S粘连蛋白启动子区,经EMT过程抑制HCC浸润与远处转移[37]。
3 KLFs与HCC诊断、预后
KLF4的表达缺失与HCC的组织学分级密切相关,是原位肝移植术术后的一个独立的阴性预后因素。KLF4的表达促进了HCC细胞的分化。HNF-6是KLF4的一个新的转录靶点,是KLF4诱导HCC分化的关键分子介质。将这一新的生物标志物整合到传统的米兰标准中,可以提高HCC的预测准确性[38]。KLF4和Sp1调控YY1-结合蛋白(RYBP)的表达,并与HCC预后相关。失调的KLF4和Sp1有助于降低HCC肿瘤组织中RYBP的表达。肿瘤中RYBP水平降低(与改变的KLF4和Sp1表达相关)与肿瘤大小、较差的分化和对远处转移的易感性增加有关,失调的KLF4、Sp1和RYBP可能导致较差的预后[39]。RYBP可能代表癌症治疗目标,并可能作为HCC的预后生物标志物,单独或与KLF4和Sp1联合使用,KLF4的表达与肿瘤分化显著相关。Ki-67增殖指数在分化良好的HCC中显著降低,但低KLF4表达与高KLF4表达之间差异无统计学意义,Kaplan-Meier分析显示,KLF4的高表达与较长的疾病特异性存活显著相关。单变量和多变量分析显示,高KLF4表达与更好的疾病特异性存活相关,是更好的疾病特异性存活的独立预测因子[40]。
KLF5在HCC和HCC细胞株标本中过表达,预示着HCC患者预后不良。miR-145-5p与KLF5有关并对HCC起作用,microRNAs(miRNAs)是一类内源性、小的非编码RNA,转录后可调控真核生物中互补靶mRNA的表达,其影响包括细胞感染、发育、免疫和致癌等多种生物学过程。miRNA表达谱表明在各种不同的癌症类型中存在大量的miRNA标记。miRNA信号可能预测癌症预后、分类和治疗反应。肿瘤干细胞具有高度的致瘤性、转移性和耐药性,导致临床患者对治疗的反应减弱、肿瘤转移和复发以及预后恶化。流式细胞术分析HCC细胞系CD44和CD133亚群,发现CD44High/CD133High和CD44Low/CD133Low细胞亚群。随后对这些亚群进行了排序,并通过使用下一代测序技术进行RNA测序,确认KLF5是在CD44High/CD44High细胞中显著上调的基因。KLF5过表达丰富了CD44High/CD133High亚群,与CD44High/CD133High细胞上调调控一致,KLF5过表达细胞对抗癌药物具有更强的耐药能力,显示更强集落形成能力。相比之下,小干扰RNA对KLF5敲除降低CD44High/CD133High亚群。当KLF5 TGF-β1乙酰化,以KLF5为媒介的CD44High/CD133High族群浓缩。与野生型KLF5相比,缺乏乙酰基KLF5突变体异位表达增加CD44High/CD133High亚群[41]。
KLF6表达在HCC组织中与邻近的非癌组织相比显著下调,且与HCC患者淋巴血管空间侵袭呈负相关[42]。此外,KLF6过表达降低细胞增殖并减弱了细胞侵袭能力,随后在HCC细胞中PCNA和MMP-9的表达降低。血小板释放通过抑制KLF6的表达促进肝癌细胞的增殖。原发性肿瘤微环境中的血小板在肿瘤进展的调节中起着至关重要的作用。血小板释放在体外和体内对HCC细胞发挥增殖作用。该作用取决于HCC细胞中KLF6表达降低。与血小板或血小板颗粒内容物孵育后,SMMC/7721和HepG2细胞表现出显著的增殖增加和凋亡减少。然而,重新悬浮活化的血小板颗粒孵育癌细胞时未观察到效果,所述活化的血小板颗粒耗尽释放物。血小板释放还增加细胞周期S期和G2/M期HCC细胞群,并减少了G0/G1期细胞群[43]。Kaplan-Meier分析显示,KLF6高表达与更好的总体存活相关。KLF6阴性患者生存率低于KLF6阳性患者。
KLF8在HCC中高度表达,并促进HCC细胞增殖和侵袭促进肿瘤起始和进展。KLF8在HCC及远处迁移组织中高表达,KLF8高表达患者预后不良。
4 KLFs在HCC中的作用机制
4.1 促进作用KLF5通过PI3K/AKT/Snail信号通路促进肝癌的EMT[44]。KLF5在HCC细胞中的沉默可下调N-钙黏蛋白、波形蛋白和Snail的表达,并增加上皮标记E-钙黏蛋白的表达。MMP2和MMP9的表达在KLF5沉默的HCC细胞中也有下降,而KLF5过表达时结果相反。CD44是调节因子CTNNB1的靶点之一,研究发现部分HCC细胞中存在调节因子CTNNB1的突变,KLF5可以通过间接调控作用调控CD44的表达,促进HCC的发生。miR-145-5p与KLF5有关并对HCC起作用,确定KLF5为miR-145-5p在HCC细胞中的靶点,miR-145-5p通过靶向KLF5降低HCC的增殖和迁移,KLF5过表达部分减弱了miR-145-5p的抑癌作用。KLF5表达与HCC组织中miR-145-5p水平呈负相关[45]。KLF5具有抑制肿瘤和促进肿瘤生长的双重作用,研究表明在培养的非癌上皮细胞中,KLF5在TGF-β诱导的乙酰化作用下从增殖活性转变为抗增殖活性,从而改变KLF5转录复合物和p15、MYC等基因的表达。KLF5的乙酰化状态在肿瘤发生中也起相反作用,使用PC-3和DU145前列腺癌细胞系[46],其增殖被TGF-β抑制。KLF5抑制这些癌细胞的增殖,并且抑制依赖于KLF5乙酰化。MYC和p15显示在非癌细胞中发现相同的表达变化模式。在裸鼠中,KLF5还以乙酰化依赖性方式抑制肿瘤生长。总之,KLF5抑制TGF-β的存在,导致KLF5的乙酰化作用,但中断KLF5乙酰化作用又对肿瘤起促进作用。中断KLF5乙酰化将其在前列腺癌细胞中的功能由抑癌因子转化为肿瘤启动子,去乙酰化将KLF5转换为肿瘤促进活性,并且阻断TGF-β信号传导减弱了KLF5的肿瘤抑制活性。
KLF8的上调通过与VEGFA启动子的CACCC区结合,增加VEGFA蛋白水平,诱导VEGFA启动子活性。此外,KLF8还调节HIF-1α和局灶性FAK的表达。PI3K/AKT抑制剂LY 294002抑制KLF8诱导的VEGFA表达,而PI3K/AKT信号通路蛋白如P-PDK1和P-AKT显著降低。KLF8高表达肝癌细胞具有较高的诱导血管生成的潜力。KLF8可能通过与VEGFA启动子的CACCC区结合,诱导VEGFA启动子活性,并通过FAK激活PI3K/AKT信号调节HIF-1α的表达而诱导肝癌血管生成[47]。
KLF10缺乏通过TGF-β/SMAD途径抑制HCC发生过程中肝细胞的增殖,转化生长因子-β敏感性的丧失被认为是HCC发展过程中的一个事件。KLF10基因激活可促进转化生长因子β易感的人HCC细胞的生长抑制和凋亡,抑制stathmin启动子活性,提示stathmin是KLF10的直接靶点。此外,还发现KLF10通过与GST-P2结合,抑制谷胱甘肽转移酶P(GST-P)启动子活性,GST-P是HCC发生过程中一个很好的肿瘤标志物。KLF10的缺乏通过促进肝细胞发生过程中的TGF-β/SMAD途径而阻断肝细胞的增殖,同时KLF10在肝硬化中高表达,关于KLF10的资料不足,在肝脏肿瘤发生中的实际作用有争议[28]。
4.2 抑癌作用KLF4和Sp1,它们分别直接结合RYBP的启动子区域以诱导和抑制RYBP转录,KLF4抑制,而Sp1通过调节RYBP表达促进HCC细胞生长[39]。另外,miR-9-5p在HCC中过表达作为KLF4的新上游基因,miR-9-5p表达与临床样品中的KLF4表达呈负相关。Kaplan-Meier分析显示,miR-9-5p高表达组中HCC的预后明显不良。miR-9-5p直接结合KLF4的3′UTR,这降低了KLF4的表达水平。miR-9-5p/KLF4轴促进HCC增殖和迁移,并抑制HCC凋亡。miR-9-5p上调Bcl-2/Bax蛋白比率并激活AKT/mTOR信号传导。miR-9-5p/KLF4轴能够促进HCC进展,其可以通过调节AKT信号传导途径发生,突出了HCC治疗中潜在的新靶标。TGF-β1通过激活miR-135a-5p而下调KLF4的表达。miR-135a-5p通过直接靶向KLF4促进HCC细胞的增殖和转移。miR-135a-5p在HCC组织表达上调,与KLF4表达呈负相关。TGF-β1通过激活miR-135a-5p而下调KLF4表达,促进HCC增殖和转移[48]。
KLF6作为一种新型肿瘤抑制基因参与多种生物学行为,在调节肿瘤细胞生长和侵袭中起重要作用。KLF6和Sp1相互作用调节basigin-2表达介导的HCC增殖、侵袭和转移[49]。肿瘤抑制基因KLF6及癌症相关分子basigin-2在HCC进展和转移中起重要作用。Sp1是SP/KLFs家族成员之一,它调节HCC中basigin-2的表达。在HCC组织中KLF6表达低而Sp1和basigin-2表达高,KLF6表达高而Sp1和basigin-2表达低。KLF6与basigin-2和Sp1启动子结合并降低其表达。KLF6可以通过与其启动子结合直接抑制basigin-2表达,或通过抑制转录因子Sp1的表达间接抑制基因表达来间接抑制basigin-2表达。KLF6的过表达通过靶向basigin-2在体外和体内抑制HCC细胞的侵袭、转移和增殖。
另外,KLF6通过调节VAV3-RAC1信号轴来抑制HCC传播,KLF6是HCC细胞迁移的一种潜在调节因子。KLF6基因敲除可促进细胞迁移,与KLF6 mRNA水平下降与人HCC血管浸润的关系一致。同时,单拷贝删除KLF6的HCC小鼠模型中,肿瘤形成增加,肺转移增加,存活率降低,说明KLF6抑制了HCC的发展和转移。结合基因表达谱和染色质免疫共沉淀深度测序,在包括已知的RAC1小GTPase激活因子VAV3在内的HCC细胞中发现了新的KLF6转录靶点。事实上,KLF6基因敲除细胞的RAC1活性以VAV3依赖的方式增加,而RAC1或VAV3的敲除则会影响HCC细胞的迁移。KLF6通过调节VAV3-RAC1信号轴来抑制HCC传播[50]。
KLF17是EMT的负调节因子,参与抑制肿瘤的转移,KLF17的低表达和失活是由肿瘤中的微RNA、基因突变和杂合性缺失引起的。KLF17的低表达增加了肿瘤的转移活性,其机制是:低KLF17通过调节EMT相关基因的表达而介导EMT,而下调的KLF17则经上调DNA结合抑制剂1(ID1)增加肿瘤转移。突变型p53蛋白与KLF17形成复合物,介导KLF17缺失,抑制EMT基因转录,增加肿瘤转移[51]。
另KLF17在增强TGF-β/SMAD信号通过SMAD3依赖通路抑制肿瘤的进展。TGF-β/SMAD3信号传导诱导KLF17表达,产生正反馈环。TGF-β/SMAD3-KLF17环是肿瘤细胞抗转移和抑瘤的关键。沉默KLF17使SMAD结合元件(SBE)上的SMAD3-DNA复合物形成,并影响TGF-β的表达[52]。KLF17改变SMAD3在染色质上结合模式,调节TGF-β/SMAD3依赖靶基因。SMAD3和KLF17通过KLF17响应元件/SBE区域相互作用。TGF-β刺激染色质上KLF17向转移相关基因亚群的募集。
5 KLFs与HCC治疗
通过KLF5对HCC细胞体外和体内增殖、凋亡、迁移和侵袭影响的研究,证明KLF5的抑制明显抑制了HCC的生长和转移,而KLF5的过表达促进了这些过程。KLF5在HCC的进展和转移中起重要作用,通过激活PI3K/AKT/Snail信号通路诱导EMT的发生,抑制KLF5可能是一种潜在的治疗方法。研究[45]表明,miRNAs在HCC的发生、发展中具有重要作用,某些抑癌miRNAs在HCC中已经被鉴定,确定KLF5为miR-145-5p在HCC细胞中的靶点。miR-145-5p通过靶向KLF5降低HCC的增殖和迁移,KLF5过表达部分减弱了miR-145-5p的抑癌作用。KLF5表达与HCC组织中miR-145-5p水平呈负相关。因此抑制KLF5是一种潜在的治疗方法。
KLF6过表达抑制了异种移植肿瘤的生长。KLF6在体外和体内抑制HCC细胞的生长和侵袭,表明KLF6具有肿瘤抑制功能。敲低KLF6表达显著减少了血小板介导的HCC生长的增强。用TGF-β受体抑制剂SB431542阻断TGF-β信号传导抵消了血小板对KLF6表达和HCC细胞增殖的影响。研究表明,血小板释放,特别是TGF-β,通过降低KLF6的表达促进SMMC.7721和HepG2细胞的增殖。因此KLF6可能用于预防和治疗HCC的潜在新治疗靶标[43]。
KLF8通过激活Wnt/β-连环蛋白信号传导途径促进干细胞样特征,在HCC发生、发展中发挥有效的致癌作用。当用相同剂量的索拉非尼或顺铂处理时,与对照细胞相比,在敲除KLF8的HCC细胞中细胞凋亡显著增加。因此,靶向KLF8可提供抑制HCC致瘤性的有效治疗方法[47]。
功能上,KLF17的缺失增强了癌细胞的致瘤特征。KLF17对TGF-1的全部细胞抑制功能至关重要。临床上,KLF17在HCC早期表达明显下降。KLF17与癌组织中SMAD3水平呈正相关。增强KLF17的活性对TGF-β的靶向治疗有重要的治疗意义,β/SMAD3通路,TGF-β/SMAD-KLF17通路在肿瘤转移过程中相互影响,为TGF-β的调控提供了新的模型。β/SMAD3信号传导系统定义了KLF17的抗转移功能[52]。
6 展望
综上所述,KLFs家族成员与多种肿瘤的发生、发展相关。已知有7种成员与HCC的关系密切,其机制是在HCC发生、发展中起抑制或促进作用,对HCC的早期诊断、治疗和预后有重要作用,具有临床应用前景。目前HCC早期诊断困难,治疗预后差,5年生存率低。因此,对KLFs家族与HCC关系的研究具有重大意义。除了这7种KLFs,KLFs家族其他因子也可能与HCC有关。KLFs家族对HCC产生作用的多个KLFs可能有交互协调作用,有待研究。
猜你喜欢
中国交通信息化(2022年8期)2022-10-28
中国生物化学与分子生物学报(2022年8期)2022-09-08
九江学院学报(自然科学版)(2022年2期)2022-07-02
保健医苑(2022年4期)2022-05-05
昆明医科大学学报(2021年10期)2021-12-02
复旦学报(自然科学版)(2021年5期)2021-11-17
汽车维修与保养(2021年8期)2021-02-16
现代临床医学(2021年1期)2021-01-26
医药前沿(2020年29期)2020-12-04
山西医科大学学报(2020年8期)2020-09-16
