
Mn-基超级电容电极材料的研究进展
2019-08-16 08:23张万英胡良胜鲁福身
汕头大学学报(自然科学版) 2019年3期
张万英,胡良胜,鲁福身
(汕头大学化学系,广东省有序结构材料的制备与应用重点实验室,广东 汕头515063)
0 引言
随着世界人口的增长和工业的发展,人们对能源的需求急剧增加,由于目前的能源主要由不可再生的化石资源提供,导致化石资源储量不断减少甚至枯竭,同时,排出大量的CO2对环境造成极大的污染和破坏.因此,科学家正在积极开发和利用可替代再生清洁能源,如太阳能、风能、潮汐能、地热能等.然而,这些能源具有不连续性的缺点,因此需要将其进行储存.在众多能源储存元件中,超级电容器由于具有功率密度高、充电时间短、使用寿命长、温度特性好、绿色环保等特点,被视为最具有潜在实际应用前景的储能器件而受到广泛关注.RuO2是一种早期典型的赝电容比较大的电极材料,但贵金属Ru的成本较高限制了其商业应用[1].Mn-基材料,MnO2的理论电容可达到1 370 F/g,但在实际研究中,因为导电性差(10-5-10-6S/cm)、循环稳定性欠缺以及负载量低等因素导致其容量远未达到理论值[2],目前最多只能达到理论的60%左右[3].但是Mn-基电极材料因为其电势窗口宽,来源丰富,环境友好和成本低等优点使其成为极具潜力的储能电极材料.因此,目前大量研究通过电极材料的结构优化[4-5]、与导电基复合[6-8]、金属掺杂[9-10]、缺陷工艺[11-12]和异质结构[13-14]等手段来提高Mn-基材料的功率密度、负载量和比电容.
1 Mn-基超级电容器的储能机理
自从Lee发现MnO2具有赝电容行为以来[15],众多课题组对Mn-基材料的法拉第反应过程和电化学性质进行了深入的研究,结果发现其储能机理包括双电层电容和法拉第电容两种电荷储存方式[16].此外,电解质的性质对于Mn-基超级电容器的电化学性能有至关重要的影响,主要体现在对电容器的电势窗口、等效串联电阻、能量密度和使用温度范围等方面[16-17].如图1所示[17],Mn-基材料在不同的介质中的法拉第电容过程截然不同.锰氧化物(MnOx)的电容主要来自于表面的氧化还原反应和体相中碱金属离子的嵌入/脱嵌.简而言之,即Mn(II)↔Mn(III)+e-.法拉第反应过程如式(1):

X代表H,Li,Na,K;MnOa(OX)b代表高价态锰,MnOa-b(OX)b+d代表低价态锰.以MnO2为例,在不同pH的电解质中,MnO2的反应过程如式(2):

在中性电解质中,电容器的充电/放电过程会使MnO2发生还原/氧化反应,伴随着其晶格间距的膨胀/收缩.同时在充电过程中,电解质中的质子经过双电层扩散到MnO2晶格表面,并与MnO2晶格表面的O2-结合生成MnOOX,随着反应时间增加质子和电子不断向MnO2晶格内部转移,直到将MnO2转化为MnOOX,完成充电过程[16].
在酸性电解质中,MnO2的还原/氧化是通过化学吸附/解吸或H+的嵌入/脱嵌来平衡(如图1)[17].然而,酸性电解质中过量的H+会使MnO2发生歧化反应(MnO2+H++e-→MnOOH;MnOOH+3H++e-→Mn2++2H2O),导致电极材料溶解而降低其稳定性.由于从Mn2+到Mn4+的氧化过电位较高,Mn的这种溶解和还原过程几乎不可逆,使得电极的倍率性能较差.当pH值在3~7之间,溶液中同时存在Mn2+和MnOOH这两种物质,随着pH值的增加,Mn2+的含量会逐渐降低[3].因此,基于以上规律,对Mn-基超级电容器电极材料在水系电解质的研究中,大多数人使用中性溶液,如Na2SO4或LiCl作为电解质,或者通过在MnO2面膜覆盖一层导电碳材料或导电聚合物,可以在一定程度上减少MnO2的溶解[18-19].
在碱性电解液中,因为大量OH-聚集在MnO2表面而使其转化为绝缘的Mn(OH)2层,覆盖了活性位点和降低了反应动力学速度.在碱性电解液中,研究者认为电极材料会发生腐蚀反应,其电化学反应被认为可能如式(3)-(5)[20-21]:

图1 氧化还原过程和MnO2在酸性、碱性和中性电解质的储能机理[17]
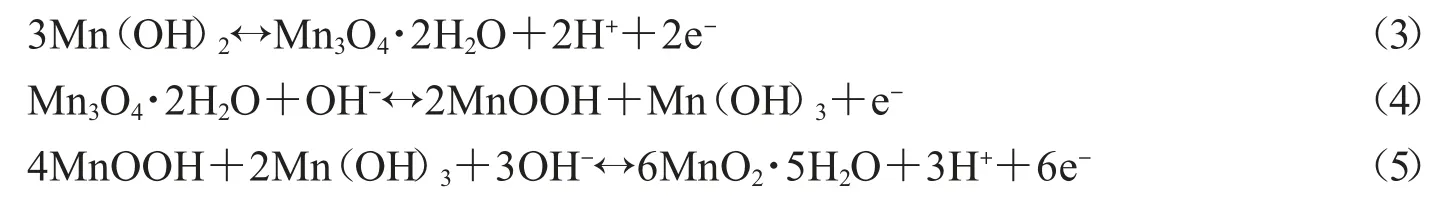
对于Mn(OH)2-基电极材料,Mn(OH)2先被氧化为MnOOH和MnO2,使得Mn(OH)2表现出赝电容行为,有关反应如式(6)-(7)[22]:

2 Mn-基材料晶体结构
MnO2都是以如图2所示的MnO6八面体为基本单元构成,其O原子位于六个角位置,Mn原子位于中心[23].由MnO6八面体基本单元之间可以沿棱边或顶点相结合而形成链,这些链通过角共享可以形成不同的键合方式从而组成不同的晶体形式.常见的MnO2晶体结构有 α-、β-、γ-、δ-和 λ-MnO2,其晶体结构如图2所示[24].虽然MnO2主要的晶体结构有5种,但可以概括为3大类:链状或隧道结构的α-、β-、γ-MnO2型;片状或层状结构的δ-MnO2;以及立体结构的λ-MnO2[25].
以δ-MnO2为例,它是由棱边共享的MnO6八面体双链组成,在四角形晶胞中连接成一维的(2×2)与(1×1)隧道,(2×2)隧道的宽度大约是4.6Å,这足够阳离子嵌入和脱嵌.然而,由单链共边MnO6八面体组成一维(1×1)隧道(约1.89Å)的β-MnO2与三维尖晶石结构的λ-MnO2,其隧道宽度小于K+直径,所以在钾盐电解质中有表现出较低的电容值.γ-MnO2由斜方锰矿(1×2)和软锰矿(1×1)隧道两种共存晶胞随机组成,在一维方向上的隧道宽度相似.而δ-MnO2为层间距约7Å的二维层状结构,它含有大量的水和稳定的阳离子,例如在MnO6八面体薄片之间的Na+或K+[23].与δ-MnO2的结构类似,Mn(OH)2具有层状晶体结构,层间距为4.7Å,这种特殊结构,不仅可以容纳大量的离子,也极大地促进了离子的扩散[26].
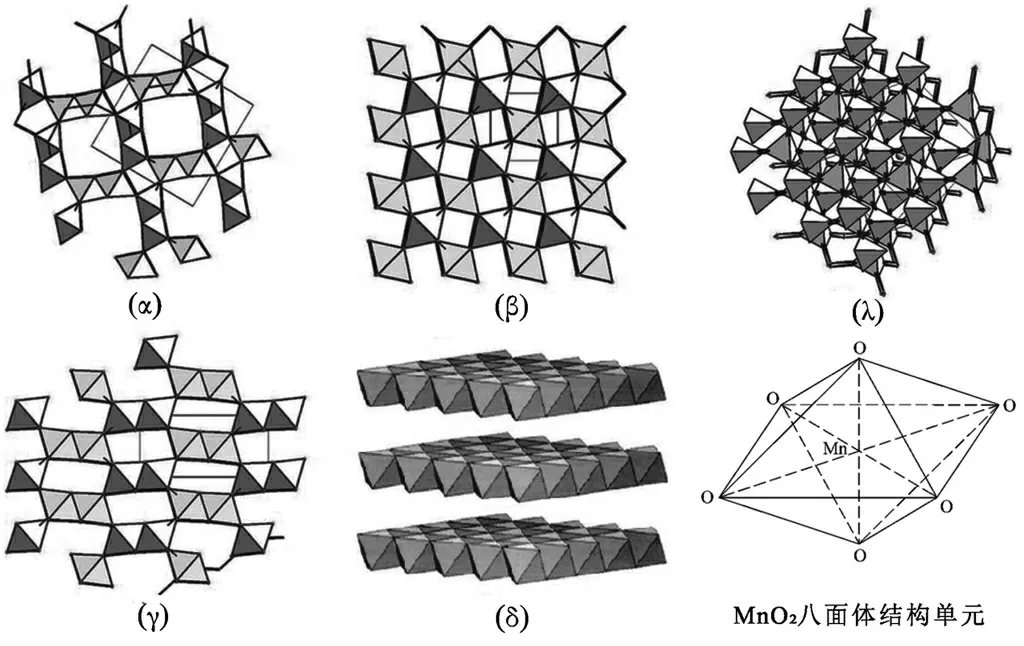
图2 α-、β-、γ-、δ-和 λ-MnO2晶体结构[24]和MnO2八面体结构示意图[23]
氧化锰的电容大小高度依赖于其晶体结构,因不同晶型的隧道的宽度有所不同,隧道的宽度影响阳离子的传输和嵌入,在MnO2的多种晶体结构中,孔隙宽度足以容纳电解质离子的结构具有更大的电容.除研究较多的MnO2电极材料之外,其他锰氧化物如MnO、Mn3O4和Mn2O3也被用来作为电容器电极材料,其晶体结构如图3所示.除电极材料本身的结构对其电容大小具有主要影响以外,电解质对体系的电容大小也有一定的影响,为了使超级电容器的比电容达到最大,根据隧道尺寸来选择最兼容的电解质也是必须考虑的一个重要因素[27].
图3 (a)岩盐结构(MnO),(b)尖晶石(Mn3O4),(c)方铁锰矿(Mn2O3)[28]
3 Mn-基超级电容器电极材料研究现状
Mn有5个未成对电子,具有较多氧化态,这种独特的电子结构使得Mn-基氧化物具有极强的氧化还原活性,因此它可以以多种不同的氧化态和不同晶体形态的氧化物的形式存在(如:MnO、Mn3O4、Mn2O3、MnO2、MnO3、Mn2O7等).其中大多数具有特殊的隧道结构,能够进行大量氧化还原反应.如MnO、Mn3O4、Mn(OH)2以及其他的Mn-基材料.目前为止,Mn-基材料应用于超级电容器的研究中最多的是MnO2.
3.1 MnO2-基电极材料
如上所述,MnO2有多种晶体结构,在超级电容器的应用中,不同晶体构型和形貌的电极材料的电化学性质有所差异.从形貌的角度来说,MnO2纳米材料作为超级电容器电极的活性材料时,影响其电化学性能的主要因素有三个:1)氧化还原活性位点的数量;2)纳米颗粒在宿主材料中的分散情况;3)纳米材料的负载量.通常材料的形貌会影响活性物种的负载量、活性位点的暴露面积以及质子和离子的转移和电子传递速度.目前报道比较多的形貌主要有四大类,即零维、一维、二维和三维纳米结构.
零维结构:与其他形貌的材料相比,零维纳米颗粒具有比表面积大,暴露活性位点多等特点,但颗粒与颗粒之间会产生较大的接触电阻致使电极的导电性降低,此外,纳米颗粒容易团聚,经过多次充放电循环后电容值急剧衰减.因此,将零维纳米颗粒与其他一维材料复合或者嵌入到合适的高导电基体材料中,不仅可以保持纳米颗粒的活性位点,还能有效克服导电性差和易团聚的问题.有研究报道,纳米颗粒的尺寸应控制在20 nm之内,如尺寸大于20 nm其比电容将急剧下降.Kiani等人报道以介孔二氧化硅为硬模板,蔗糖为碳源制备有序介孔碳前驱体(CMK-3),然后将Mn前驱体浸渍在CMK-3中,通过退火法制备出不同比例MnO2/CMK-3复合材料,如图4(a)和4(b)所示.当MnO2质量分数达到40%时,复合材料的电容性能最佳,进一步增加MnO2含量,由于会导致颗粒严重团聚而使其容量降低.基于质量分数40%的MnO2/CMK-3复合电极组装成对称超级电容器时其比电容为640 F/g(1 A/g,2.0 V,0.5 mol/L Na2SO4),经过10 000次循环后,比电容几乎没有衰减[29].
一维:一维形貌的MnO2纳米结构主要有纳米棒、纳米纤维和纳米线等.在这些结构中,由于纳米线的横纵比大,可以减小离子扩散距离,因而有更好的离子扩散率和导电性.Xu等人报道了一种选择性沉积生长工艺,在磁场的辅助下制备垂直排列的超长镍纳米线阵列(NNA),如图4(c),长度可达1 mm.在纵横比超过8 000的高表面积的导电基体NNA上沉积一层无定形的MnO2,如图4(d),与其他材料相比,在相同的MnO2负载量下,NNA使活性材料的厚度变薄,使得MnO2表面积增大而暴露更多的活性位点,并且与电解液能够充分接触.得益于这独特的结构,使其表现出极大的面电容(750 mF/cm2,1 mV/s,0.5 mol/L Na2SO4)[30].
二维:二维的MnO2(如纳米薄片、纳米薄膜),由于具有大面积暴露的氧化还原活性位点而被研究的最多.例如Wang等人先通过在泡沫铜上制备出无定型的纳米碳材料,以此为模板与KMnO4原位发生反应,获得以超薄κ-水钠锰矿类石墨烯状MnO2堆积而成的海绵状结构,如图4(e)和(f)所示.该海绵状结构MnO2的负载量高达4 mg/cm2,比表面积达306.9 m2/g,在负载量大的基础上同时暴露出大量的氧化还原活性位点,使其电容值达516.7 F/g(0.2 A/g,1 mol/L Na2SO4).此外,此电极材料还有极好的循环能力,循环10 000次后,电容增加到129%(10 A/g),这是因为电解质最初只与电极材料最表层接触,随着时间的延长,接触到更多的材料,部分最初未与电解质接触的材料在循环过程中慢慢也参与氧化还原反应,从而表现出电容值增大的趋势[31].
三维:与二维纳米结构相比,三维纳米结构电极通过合理的形貌和结构设计,可以与电解质离子更好的接触.例如,介孔结构具有较高的表面积和均匀的孔径,是超级电容器电极应用的首选材料.Bag等人利用模板法合成具有大表面积(238 m2/g)的介孔δ-MnO2三维结构,如图4(g)-(h).作者研究发现,当平均孔径为3.6 nm时,有利于电荷存储过程中电荷转移/离子扩散,其电容大小为364 F/g(1 A/g,0.5 mol/L Na2SO4).此外,该多孔结构的稳定性较好,经过3 000次循环后电容值几乎没有衰减.活性材料的尺寸大小对电容值影响非常明显,当MnO2簇大于200 nm时,MnO2的氧化还原活性位点与集流体之间的接触受到阻碍,从而降低了电荷的运输和转移[32].

图4 (a)-(b)MnO2/CMK-3的TEM图[29],(c)-(d)分别为NNA和NNA@MnO2的TEM图[13],(e)-(f)分别为石墨烯状MnO2的TEM图和高倍TEM图[31],(g)-(h)分别为介孔δ-MnO2的TEM和FESEM图[32],(b)和(e)图中插图为其对应的选区电子衍射
3.2 MnO-基电极材料
在各种Mn-基材料中,MnO-基电极材料电容值比较高,当它组装成不对称超级电容器时,能量密度和功率密度高于大部分MnO2-基材料[37].在已有的报道中,MnO-基能量密度已达到59.6 Wh/kg(功率密度为1 529.8 W/kg)[34].MnO是典型的传统电池材料,其电化学性能优异,近年来,有不少的研究者把MnO纳米结构材料应用到超级电容电极材料中,也获得比较理想的结果.但在合成MnO的过程中几乎都要经过高温煅烧或退火等热处理,合成材料的形貌大多以纳米颗粒为主[33-36],所以单一的MnO物种易团聚从而导致活性位点少和稳定性差等问题.因此,研究者们通过设计将MnO与导电基体结合形成复合结构来克服其易团聚的问题.
合成MnO纳米颗粒一般是将有二价锰有机化物与导电基体混合,再经过热处理制得.例如,Li等人采用油酸锰、油酸和十八烯为前驱物,将其充分混合并烘干后进行高温热解,合成平均粒径为22.5 nm的单分散球形MnO纳米晶.该纳米晶具有良好的电化学性能和稳定性,比电容高达736.4 F/g(1 A/g,1 mol/L Na2SO4),经过5 000次循环后比电容保持93.3%.此外,以活性碳(负极)与MnO纳米晶组成不对称超级电容器,该装置的能量密度为44.2 Wh/kg(功率密度为900 W/kg)[34].最近,Zhang等人通过把干木耳浸泡在1 mol/L乙酸锰溶液,木耳的尺寸在浸泡前如图5(c),浸泡后图5(d).作者利用木耳的多孔性质和富含几丁质(Chitin)的特点充分吸附Mn2+,再将其在惰性气氛800℃下煅烧,将Mn2+转化成MnO的同时木耳碳化成生物质多孔碳(BPC),从而制备出MnO/BPC复合材料,如图5(a).木耳外层的纤维素层可通过物理吸附少量的Mn2+,内层丰富的几丁质可以与Mn2+发生络合反应而均匀吸附大量的Mn2+(图5(b)),因此在热处理过程中会形成两种不同尺寸的纳米颗粒,如图5(e)和5(f).由于几丁质与Mn2+发生络合反应可认为是化学吸附,Mn的负载量高,且分布均匀.因此MnO/BPC复合材料MnO负载量达75%,且内部的MnO颗粒均匀分布在碳基体中,有效防止其团聚.此复合材料具有大的比表面积、高效的电子转移通道和优异的电解质润湿性,因此具有非常高的电容735 mF/cm2(3 mA/cm2,3 mol/L KOH).另外,与活性碳(负极)组装成不对称超级电容器时,在800 W/kg功率密度下具有35.9 Wh/kg的比能量,其优异的电化学性能主要是BPC导电碳基不仅为MnO颗粒提供高导电性的骨架而便于电子传输,还为MnO在碱性电解液的腐蚀性提供保护层[33].

图5 (a)MnO/BPC合成示意图,(b)几丁质与Mn2+络合结构图,(c)-(d)木耳浸泡前后变化,(e)Mn2+络合形成的MnO颗粒,(f)纤维吸附形成的MnO颗粒[33]
除形貌为纳米颗粒的MnO外,也有少量报道三维结构的纳米阵列和三维网状结构的MnO[38].如Yu等人以KMnO4为前驱体,采用水热法在导电碳布上生长MnO2/CC纳米阵列,以蔗糖为前驱物通过水热法在MnO2表面包覆一层碳保护层,再经过高温热处理使MnO2发生相变制备出MnO@/CC纳米阵列.MnO直接生长在碳布上,避免使用粘合剂而引入的接触电阻,提高电极的导电性.MnO表面的碳层不仅抑制MnO2转变为MnO导致结构坍塌而保持结构的稳定,还保护活性材料免受碱性电解液的侵蚀,进而提高其稳定性.此外,通过控制热处理温度可以使MnO2发生相变得到不同的产物,在相似的形貌前提下,发现MnO的电化学性能比Mn3O4和δ-MnO2要好.以3 mol/L的KOH为电解质,在电流密度为3.7 A/g或4 mA/cm2时,其比电容为662.9 F/g或716 mF/cm2.当分别以Co3O4纳米片和MnO@C纳米片为正极和负极,组装成柔性非对称超级电容器,在1.7V的工作电势下,比电容为166 F/g,能量密度达59.6 Wh/kg[35].
3.3 Mn3O4基电极材料
Mn3O4在常温下只有单一的黑锰矿结构,但微观结构可控,是作为超级电容器潜在的电极材料之一.已报道的Mn3O4基形貌较丰富,有纳米颗粒[39]、纳米棒[40]、纳米片[41]和薄膜[42]等,相关研究主要集中在形貌对其电化学的影响,通过不同的合成方法合成不同的形貌.根据合成过程可大致分为原位合成法和分步合成法.化学浴和电沉积是最典型的Mn3O4原位合成方法,也是合成二维结构常用方法.在化学浴法中,OH-对Mn3O4的纳米结构和形貌有决定作用.例如,Zhu等人利用化学浴沉淀法自组装合成Mn3O4薄片,通过改变络合剂HMT(环六亚甲基四胺)的浓度,可以非常容易地调节堆叠纳米片层的尺寸和晶体性质,因为HMT水解产生的OH-决定Mn3O4的生成速率,随着OH-含量增加,由小颗粒自组装的Mn3O4形貌从不规则变为规则(图6).因此,低浓度的HMT合成的样品比表面积大(104 m2/g),孔径分布均匀,结晶度低,以致比电容高达398 F/g(5 mV/s,1 mol/L Na2SO4)[42].Aghazade等人采用电沉积法,在不锈钢基体制备了纳米片-纳米棒分级的Mn3O4纳米结构,可能是因为阴极表面H2鼓泡中断了粒子的横向生长,使得纳米棒的直径为50 nm,而长度长达300 nm,并且由大部分的纳米棒组装成二级的片状结构.得益于这独特的结构,其比电容达279 F/g(2 mA/cm2,1 mol/L Na2SO)4[40].在以碳材料为导电基体制备的合成中,大部分都是两步合成,石墨烯是合成Mn3O4-基最常用的导电基体[43-45].例如,Dubal等人先用微波等离子体增强化学气相沉积法在碳纤维上制备出垂直排列的石墨烯纳米薄片(VAGN/CF),再通过水热法在VAGN/CF上生长Mn3O4纳米颗粒,独特的三维纳米阵列结构使该电极比容高达670 F/g(0.7A/g,1 mol/L Na2SO4).当组装为全固态对称超级电容器时,该装置的电容达562 F/g,能量密度为50 Wh/kg(功率密度为64 kW/kg),经过10 000次循环后电容容量基本没有衰减.同时,由于碳纤维具有柔韧性,在弯曲至150°后其电容性能没有发生任何变化,该电容器表现出优异的柔韧性[41].Zhu等人首先采用改进的Hummers方法,以KMnO4为氧化剂和锰源制备出GO/MnO2纳米复合材料,再将GO/MnO2在400℃退火同时使GO还原成还原态石墨烯(rGO)和MnO2发生转晶得到rGO/Mn3O4复合材料,Mn3O4纳米颗粒均匀地锚定在rGO薄片上,Mn3O4的含量达70.7%.该复合材料的比电容为271.5F/g(0.1 A/g,6 mol/L KOH),此外,在10 A/g的高电流密度下,经过20 000次循环后电容保持率基本不变[46].

图6 自组装合成Mn3O4薄片示意图[42]
3.4 Mn(OH)2基电极材料
由于Mn(OH)2与δ-MnO2有类似的层状晶体结构,这种结构不仅可以容纳大量的离子,也极有利于离子的扩散和传输,因此,Mn(OH)2近几年也获得关注.相对于氧化锰来说,Mn(OH)2材料应用于超级电容器的报道较少,合成方法还比较单一,最常用的是电沉积法,材料的形貌主要有纳米片[47-48]、纳米薄膜[49-50]、核壳结构[51]和纳米颗[52]等.
电沉积法制备Mn(OH)2电极材料过程中,电解液在阴极发生析氢反应,使电极附近产生大量的OH-,溶液中的Mn2+与OH-在基板上形成Mn(OH)2沉淀.一般以Mn(NO3)2为电解质,在阳极同时可以制备出MnO2,例如Nayak等人用电沉积方法同时制备了两种以不锈钢为基底的MnO2/SS(阳极)和Mn(OH)2/SS(阴极)纳米片,其比电容分别为208 F/g和185 F/g(0.5 mA/cm2,1 mol/L Na2SO4)[48].Guo等人采用电沉积法,通过原位生长法在泡沫Ni上制备了二级多孔Ni纳米结构泡沫基(SPNiNF),然后在SPNiNF基体上电沉积Mn(OH)2薄膜,直接用作无粘结剂电极.当电流密度为0.5 A/g时,Mn(OH)2@SPNiNF电极具有532.7 F/g高的质量比电容和906 mF/cm2的面积电容.当与活性碳组成非对称超级电容器时,功率密度为0.6 kW/kg,能量密度69.1 Wh/kg[22].通过改变电沉积条件可以控制Mn(OH)2的形貌,Zhen等人报道通过改变电解液组成和沉积电流密度来调节,进而控制析出H2气泡的数量和大小使电极附近溶液的酸碱性发生改变,H2气泡的大小和析出速率还充当动态模板,在石墨烯上可控电沉积合成不同形貌的Mn(OH)2薄膜.在当电流密度为1 mA/cm2时,该电容器容量高达493F/g[49].
除了常用的电沉积法,也有人尝试水热法[26]、超声[52]和其他技术制备纳米Mn(OH)2.例如,Liu等人采用简单的喷涂方法,在柔性氧化铟/聚对苯二甲酸乙二酯基板上进行喷涂,制备了Mn(OH)2(/MWCNT)多壁碳纳米管复合薄膜.在扫描速率为20 mV/s下,其比电容为297.5 F/g[50].Guan等人采用一种简单的溶剂法在八面体Cu2O上直接生长超薄的Mn(OH)2纳米片,独特的结构不仅提供了便利的离子输运通道而且有利于电解质与活性物质的充分接触,使其比电容高达540.9 F/g(1 A/g,2 mol/L KOH)[51].
3.5 其他Mn-基电极材料
在锰基超级电容电极材料中,研究得比较多的除了氧化锰和氢氧化锰外,还有关于Mn2O3[53-54]、磷酸锰[55]、硫化锰[56]、碳酸锰[57]以及锰与其他金属复合[58]等的研究报道.Chee等人采用声化学法制备了Mn3(PO4)2颗粒,然后将其与聚苯胺(PANI)混合形成PANIMn3(PO4)2复合材料.Mn3(PO4)2牢固地锚定在具有支链结构的聚苯胺上,有利于电荷的快速转移,同时,由于暴露氧化还原活性位点的增加以及导电PANI与Mn3(PO4)2之间的协同增强作用,在1 mol/L的KOH电解质中,其比容量达347 F/g[55].余晨辉等人通过水热反应法在泡沫镍上生长了具有尖晶石结构的锌钴锰多元金属氧化物纳米线,在电流密度为2 A/g时其比电容量可高达1 142 F/g,当电流密度增大至16 A/g时比电容量还保持956 F/g.此电极材料具有电容高的优点,但其工作电势宽口较窄(0-0.5 V),此外,稳定性也不够理想,经2 000次充放电循环后比电容量仅剩下初始值的86%[58].
4 Mn-基超级电容器电极材料的改性
目前对于Mn-基超级电容器电极材料的改性研究中,主要是通过减小材料的尺寸来提高比表面积和暴露更多的活性位点、以及与高导电性材料(如导电聚合物、纳米碳材料等)复合来提高电极的总体导电性.此外,还可以通过精确地调整内部电子或空位结构来提高活性位点的途径而增强材料的电容性能.因此,研究者主要是从构筑异质结构、引入缺陷和金属掺杂等方法来改善Mn-基材料的电化学性质.
4.1 异质结构设计
研究报道表明由化学相容性良好的两种物质组成的异质结构,由于其具有调节电子结构和提高离子输运和电子转移作用,可以极大提高电容性能,因此,可以通过对异质结构的活性物质进行组合优化,使电化学性能得到提高[59].由材料的晶体结构与电容性能关系可知,α-MnO2和δ-MnO2的比电容值高于β-MnO2和γ-MnO2,因此,朱等人由两步水热法在α-MnO2纳米线上生长δ-MnO2纳米片获得如图7(c)-(d)所示的阵列异质结构复合材料.与纯α-MnO2(10 m2/g)相比,α-MnO2与δ-MnO2之间的协同作用提供了更大的表面积(60 m2/g)以便于快速氧化还原反应动力学的表面离子传输,并且复合结构的负载量高达3.77 mg/cm2.与纯α-MnO2比电容相比,α-MnO2/δ-MnO2的比电容提高了接近7倍,达178 F/g,面积电容为783 mF/cm2(5 mV/s,1 mol/L LiCl),经20 000次循环后的电容还保留86%[60].Liao等人首先在镍片基底上制备了垂直排列的石墨烯纳米片,再通过水热反应,将纳米MnO颗粒沉积在VAGN上,得到了MnO形貌和负载量不同的MnO/VAGN异质结构复合材料.当MnO的质量分数为37%时,MnO/VAGN电极的比电容最大,为790 F/g(2 mV/s,1 mol/L Na2SO4).而进一步提高MnO的质量分数到80%时,该电极的比电容反而减小到381 F/g,这是由于过多的MnO负载,导致部分MnO与电解质接触的机会降低和电极的总体导电性能大大降低[61].

图7 (a)α-MnO2在碳布生长示意图及晶体结构,(b)δ-MnO2在α-MnO2纳米线上生长示意图及晶体结构,(c)-(d)α-MnO2/δ-MnO2异质结的SEM图[60]
4.2 缺陷工艺
由于引入阴离子或阳离子空位可以有效地调节金属氧化物的电子结构,从而促进电极表面氧化还原反应动力学,同时还能提供额外的离子插入位点.因此,近年来,有大量的研究报道通过引入缺陷的方式来有效地提高比电容.Yang等人以尿素、蔗糖和醋酸锰为前驱物,制备含锰凝胶,经过碳化和二次沉积合成了N掺杂碳纳米片镶嵌的超小尺寸MnO纳米颗粒(直径为2-4 nm)复合结构(MnO@NCs).由于氮的强电负性使其孤对电子与相邻碳原子的p电子杂化,改变碳载体的局部几何结构和电子结构,从而优先使MnO纳米颗粒在N位成核.因为MnO纳米颗粒主要负载在N的位置,使得纳米碳片产生更多的缺陷而增强碳材料的亲水性进而增加电极与电解质溶液的接触几率.同时,由于超小尺寸的MnO纳米粒子提供了较大的氧化还原反应的活性表面,极大程度上提高了质量电容(570 F/g,2 A/g,1 mol/L Na2SO4).由于纳米颗粒均匀分散在碳层内以及纳米颗粒之间的适当距离,有效防止颗粒团聚,如图8所示,即使电流密度为5 A/g,经过6 000循环后电容仍能保留99%[62].Fu等人以MnCO3为模板剂,通过KMnO4的还原并在H2/Ar热处理,制备了一种表层具有氧空位(Ov)的MnO2而内部完整的MnO2的蛋黄-蛋壳(yolk-shell)结构(MnO2@Ov-MnO2).在1 A/g和50 A/g电流密度下,电极的比电容分别为452.4 F/g和316.1 F/g.此外,电极的稳定性特别好,在10 000次循环后仍保持92.2%的电容.MnO2@Ov-MnO2的优良电化学性能主要归因于独特的蛋黄-蛋壳、晶格中的氧空位以及复合体中两组分的协同作用[63].

图8 MnO@NCs合成示意图[62]
4.3 金属掺杂
金属(如Au,Ag,Ce,Al,Cu,Mg,Co等)掺杂可以通过调制晶体结构或物理化学相互作用,通常不仅可以增强Mn-基材料的固有电导率,掺杂原子还可以作为电子供体调节Mn-基材料的电子结构,从而获得更好的电容性能.Hu等人报道了通过水热法制备Al掺杂的MnO2,随着Al3+与Mn2+浓度比值的增加,MnO2的形貌逐渐发生变化,由直径为2μm左右的海胆状(图9(a))逐渐转变为微球状(图9(b)),最终形成0.3~3μm的不规则微球(图9(c)).由第一性原理计算研究导电性发现,与纯MnO2相比,Al掺杂的MnO2的费米能级更接近其导带最小值(CBM),因此改变材料的电化学性质,同时发现在CBM和价带最大值(VBM)之间出现了掺杂能级,减小了MnO2的禁带宽度(图9(e)),因此有效地提高MnO2的导电性.此电极材料的质量比电容可达213 F/g(0.1 A/g,0.5 mol/L Na2SO4),在循环15 000次后的电容保持率为91%[64].通过贵金属掺杂,其电化学性能更好,但贵金属成本高.例如,Kang等人通过交替沉积Au原子和MnO2来合成Au掺杂的MnO2,其电容高达626 F/g(5 mV/s,2 mol/L Li2SO4),比纯MnO2高65%[65].相比于MnO2,Mn3O4的锰和氧的键合作用相对较弱,因此更容易实现在其晶格中掺入其它元素从而极大地改善其性能.张子瑜等人报道Ce掺杂不会改变Mn3O4的晶型,但对其形貌和电化学性能有显著影响,Ce占金属离子总量的3%时的Mn3O4具有独特片状和颗粒二元结构形貌,并带有大量均匀的微孔间隙,其比电容高达477 F/g(50 mV/s,3 mol/L KOH),较未掺杂的Mn3O4提高了43%[66].金属掺杂不仅能提高电容,其稳定性也大大提高,例如Zhao等人以Sn掺杂Mn3O4/C纳米复合材料,Sn掺杂的Mn3O4/C纳米复合材料循环10 000后电容约为初始值的93%(5 A/g,6 mol/L KOH)[67].
5 总结及展望
与早期的研究相比,Mn-基材料作为超级电容器的研究近年来取得极大进展,电极的比电容和稳定性有非常大的提高.在过去的十年中,通过优化合成方法、设计形貌、制备复合材料等手段使得Mn-基材料电化学性能有了显著提高,其中MnO2研究得最多取得的成果最显著,比电容已达理论电容60%以上.在目前取得的成果中,电容性能和能量密度最优异的是MnO2和MnO,稳定性能最高的是MnO2和Mn3O4.
虽然目前的研究已取得阶段性的成果,但仍存在活性物质负载量较少、倍率性能低以及结构不稳定等问题,其限制了Mn-基材料在实际中的应用.锰基材料作为超级电容器的研究仍需朝着成本低、合成方法简单、稳定性好、功率密度大和比电容高的方向努力,以实现在实际中的应用.作为理想的锰基电容器电极材料,其应满足以下条件:
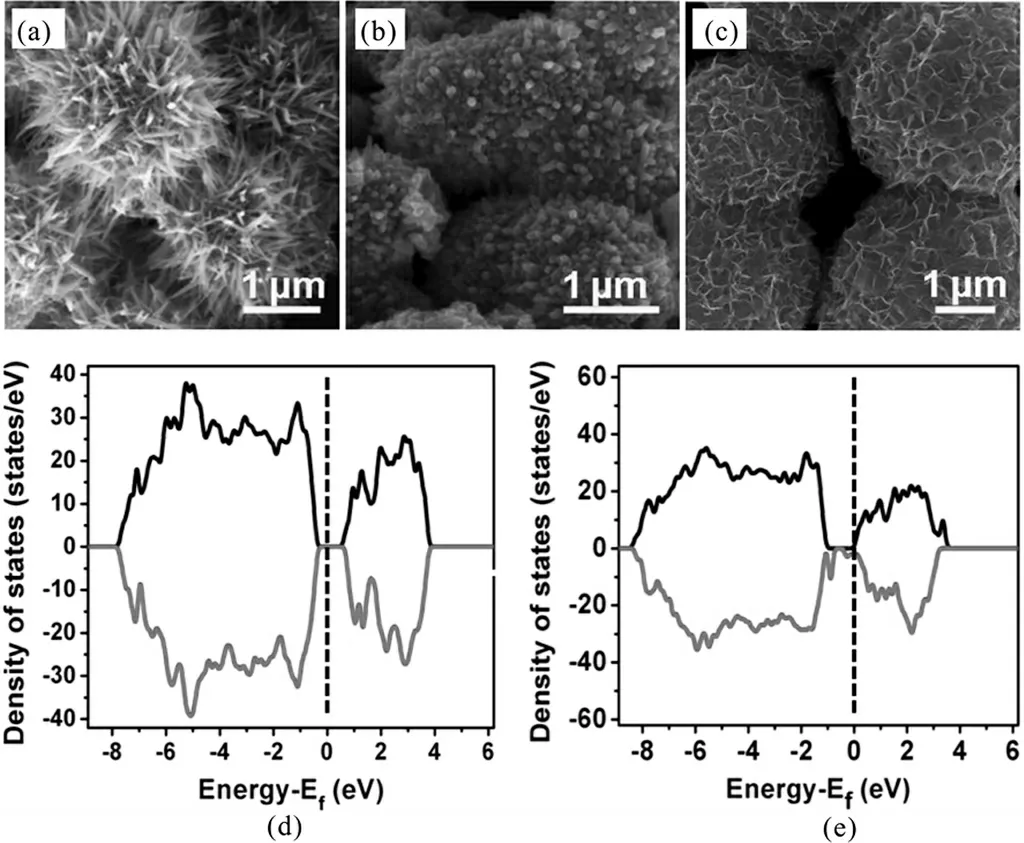
图9 (a)-(c)Al掺杂的MnO2随Al3+浓度形貌变化的SEM图,(d)-(e)分别为纯MnO2和Al掺杂MnO2的DOS图
(1)Mn-基材料的结构重复单元应细化到20 nm以内,从而最大限度地提高氧化还原反应的比表面积和反应活性中心数目;
(2)Mn-基材料应是均匀生长在导电基体表面,或通过基体改性设计出既含有活性材料又含有导电成分的纳米复合材料,以提高导电性和稳定性;
(3)Mn-基材料的形貌、尺寸和孔径分布应均匀分散,以加强电解质与活性位点的有效接触,加快离子和电子在氧化还原中快速转移.
猜你喜欢
陶瓷学报(2021年5期)2021-11-22
粉末冶金技术(2021年3期)2021-07-28
陶瓷学报(2021年1期)2021-04-13
粉末冶金技术(2021年1期)2021-03-29
物理之友(2020年12期)2020-07-16
电子制作(2019年22期)2020-01-14
中学生理科应试(2019年3期)2019-07-08
中成药(2018年5期)2018-06-06
新高考·高一物理(2017年7期)2018-03-06
中学生数理化·高二版(2016年10期)2016-12-24
