
Cirrhosis complicating Shwachman-Diamond syndrome:A case report
2019-08-14 09:22SandraCamachoLucilleMcLoughlinMichaelNowicki
World Journal of Clinical Cases 2019年12期
Sandra M Camacho,Lucille McLoughlin,Michael J Nowicki
Abstract
Key words: Shwachman-Diamond syndrome; Cirrhosis; Liver dysfunction; Case report
INTRODUCTION
Shwachman-Diamond syndrome (SDS) is a multi-organ,autosomal recessive disorder caused by compound heterozygous or homozygous mutations in SBDS gene located on chromosome 7q11[1].Common features of SDS include exocrine pancreatic insufficiency (EPI),diarrhea,failure to thrive during infancy,normal sweat electrolytes,neutropenia associated with bone marrow hypoplasia,anemia,and elevation in fetal hemoglobin[2,3].Hepatic involvement is also seen,most commonly presenting as mild elevation in transaminase levels and/or hepatomegaly.Rarely,more severe hepatic involvement has been reported.We describe a child with failure to thrive due to SDS who developed cirrhosis.
CASE PRESENTATION
Clinical observations
A 16-month-old white male presented with a chief complaint of long-standing failure to thrive first noted around 2-mo of age.The parents reported that initially he had poor weight gain but after several months he also developed poor linear growth.He maintained excellent oral intake without vomiting.Bowel movements were described as large,foul-smelling,non-watery and without blood,occurring 3 to 5 times per day.Past medical history revealed the he was delivered via spontaneous vaginal birth following a benign pregnancy and labor.Intrauterine growth restriction was not present.He had no history of recurrent gastrointestinal or sino-pulmonary infections.At presentation he weighed 6.99-kg (50% for a 4-month old infant).There were no cardiac murmurs,the lung examination revealed good air exchange without adventitious sounds.Abdominal examination revealed hepatomegaly without splenomegaly.He had markedly thin extremities with muscle wasting but no edema or clubbing.Laboratory evaluation included a complete blood count,complete metabolic panel,creatinine kinase,thyroid stimulating hormone,free-T4,serum IgA,tissue transglutaminase,and urinalysis,which were all normal except for very mild elevation of the transaminase levels (ALT = 52 U/L,ULN = 41 U/L; AST = 124 U/L,ULN = 40 U/L).Fecal qualitative fat was increased.Sweat chloride testing was normal on two occasions.Due to concern for SDS a skeletal survey was performed revealing abnormal tubulation of the long bones and narrowing of the sacral sciatic notches,suggestive for SDS.Esophagastroduodenoscopy (EGD) with biopsies and pancreatic stimulation test was performed.Biopsies of the antrum and duodenum were normal.Pancreatic stimulation test showed a generalized deficiency in pancreatic enzyme activities (trypsin,amylase,lipase,and chymotrypsinogen).Pancreatic enzyme replacement was started.Subsequent gene sequencing confirmed two heterozygous mutations in the SBDS gene confirming the diagnosis.
The patient maintained mildly elevated transaminases (Figure 1).Due to worsening hepatomegaly,and development of splenomegaly,a liver biopsy was performed at 32 mo of age.The biopsy showed hepatocytes with moderate to severe macrovesicular steatosis without necrosis and hepatocyte nodules surrounded by fibrous septa with moderate lymphocytic inflammation consistent with cirrhosis.Further testing for an etiology of cirrhosis was non-diagnostic,including ceruloplasmin,alpha-1-antitrypsin phenotype,autoimmune markers (anti-smooth muscle antibody,anti-liver-kidneymicrosomal antibody,and anti-nuclear antibody),carbohydrate deficient transferrin,and the EGL Genetic Cholestasis Panel (a proprietary test that screens for 66 genetic disorders associated with liver disease).
At 5-years of age he was noted to have pancytopenia (hemoglobin = 10.3 g/dL,white blood cell count = 1.5K/cmm,platelet count = 29 K/cmm).Examination revealed mild hepatomegaly and massively enlarged spleen.Bone marrow biopsy showed marrow hypercellularity and erythroid hyperplasia consistent with hypersplenism.He underwent an open splenectomy with normalization of his blood indices.
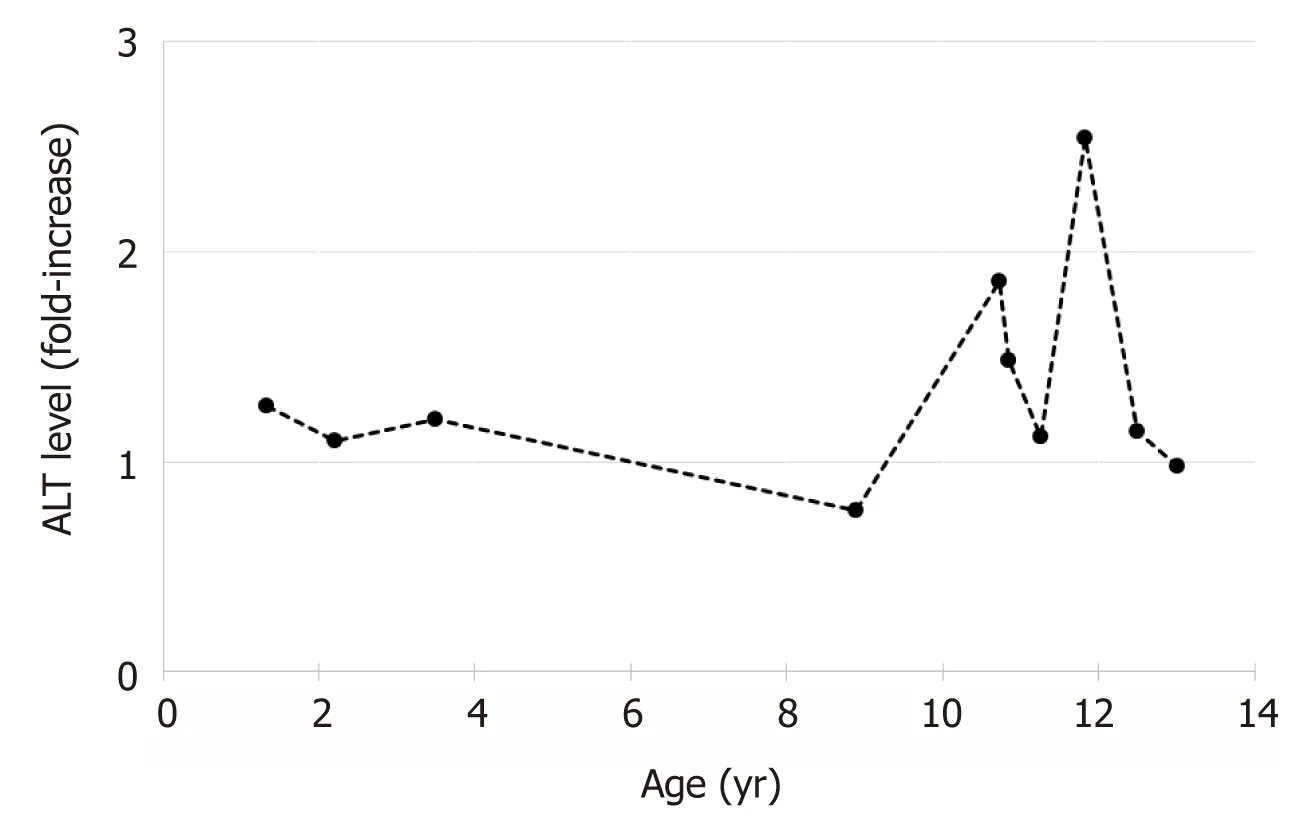
Figure1 Changes in alanine aminotransferase levels with age.
At 8-years of age,he presented with acute onset of hematemesis and hematochezia.Laboratory studies showed severe acute blood loss anemia (hemoglobin = 4.6 gm/dL); coagulation studies (prothrombin time and partial thromboplastin time)were normal.An EGD was performed revealing four large tortuous esophageal varices with wale signs and two varices in the gastric cardia; the esophageal varices were ablated with banding.The gastric mucosa showed changes consistent with hypertensive gastropathy.
At 13-years of age he remains asymptomatic,without abdominal pain,diarrhea,jaundice,pruritus,easy bruising,or gastrointestinal bleeding.Despite compliance with pancreatic enzyme replacement,his growth remains poor,with a height at the first percentile and a BMI at the second percentile for age and gender.He has no hepatomegaly or cutaneous stigmata of chronic liver disease,to include spider angiomata,palmar erythema,and xanthomas.
FINAL DIAGNOSES
The final diagnosis of the patient was Shwachman-Diamond syndrome complicated by cirrhosis,portal hypertension and resulting esophageal varices.
TREATMENT
The patient was treated with pancreatic enzyme replacement for pancreatic insufficiency due to Shwachman-Diamond syndrome.The esophageal varices were treated with band ligation without further bleeding.
OUTCOME AND FOLLOW-UP
The patient continues to have need for pancreatic enzyme replacement.He has been referred for evaluation for liver transplantation due to cirrhosis.
DISCUSSION
Typical clinical features of SDS include EPI,neutropenia associated with bone marrow hypoplasia,anemia,skeletal abnormalities,diarrhea,and poor growth;hepatic involvement is less commonly reported[1,3].The spectrum of liver involvement associated with SDS includes asymptomatic elevation of serum transaminase levels,hepatomegaly,fatty-infiltration,and varying degrees of hepatic fibrosis,including cirrhosis.
Biochemical hepatic abnormalities are a common finding in SDS patients,but are limited to elevation in aminotransferase levels; serum bilirubin,alkaline phosphatase,and gamma glutamyltransferase levels are typically normal[2,4-11].There are five studies that have assessed liver involvement in nearly 140 SDS patients,the overall incidence of elevated aminotransferase levels in these studies ranged from 57% to 100%[2,7,9-11].Mild aminotransferase elevation (< 5X the upper limit of normal) was reported in 38%to 84% of patients[2,9,10].In patients followed longitudinally,normalization of transaminase levels was seen in 56% to 67%,improvement in 33%,and no significant change in 11%[2,9].Three studies reported improvement in aminotransferase levels with increasing age[7,10,11].Mack,et al[7],reported that transaminase levels were highest before 2 years of age and normalized over time[10].Similarly,Toivianen-Salo,et al[11],reported that transaminase levels were highest in early childhood and normalized by 5-years of age[11].A pattern the mirrors the gradual improvement in pancreatic function seen in patients with SDS.
Hepatomegaly is also a well described finding in SDS,reported in 4% to 62% of affected patients[2,7,8,10,11].The liver tends to be mildly enlarged,although massive hepatomegaly can be seen[7].Hepatomegaly is a more common finding in younger children and tends to normalize in most by 3-years of age (85%-86%),similar to the pattern seen for aminotransferase levels[2,11].Histologic abnormalities on liver biopsy include varying degrees and combinations of steatosis,cellular inflammation,and fibrosis.Patients with hepatomegaly show the same spectrum of histologic findings as those without hepatomegaly[8,10].Steatosis has been reported as microvesicular,macrovesicular,and mixed micro- and macrovesicular.Hepatic steatosis is thought to arise secondary to malnutrition or infection[2].Inflammatory cell infiltrate tends to be mild and localized to the portal and periportal areas.Scarring is frequently reported as portal or periportal fibrosis,and less commonly bridging fibrosis.Cirrhosis has rarely been reported in association SDS,with no reports in over 50 years[4,5].In a review by Bodianet al[4],five children were described with SDS and cirrhosis,which was discovered at autopsy in all cases[4].Four of the children were between the ages of 12 years and 14 years,the other child was only 2 years old,and was described as having “early cirrhosis”.
The pancreas and liver share a common embryonic origin and precursor cells of these organs may share many phenotypical and functional traits,which may help explain why pancreatic and hepatic function both improve over time in patients with SDS[11].However,the mechanism of improvement remains unclear.
CONCLUSION
We report a child with SDS discovered to have cirrhosis at an extremely early age;extensive investigation failed to reveal an etiology other than SDS.It is important for Pediatric health care providers to recognize that children with SDS may have hepatic involvement which tends to be mild and improves or resolves with age.Rarely,more severe hepatic disease can occur that will require evaluation by a Pediatric Gastroenterologist.This information may be beneficial to the primary care provider in the care of children with SDS.
 World Journal of Clinical Cases2019年12期
World Journal of Clinical Cases2019年12期
- World Journal of Clinical Cases的其它文章
- Biomarkers vs imaging in the early detection of hepatocellular carcinoma and prognosis
- Study on gene expression patterns and functional pathways of peripheral blood monocytes reveals potential molecular mechanism of surgical treatment for periodontitis
- Feasibility of prostatectomy without prostate biopsy in the era of new imaging technology and minimally invasive techniques
- Safety and efficacy of transfemoral intrahepatic portosystemic shunt for portal hypertension:A single-center retrospective study
- Impact of gastroesophageal reflux disease on the quality of life of Polish patients
- Non-albicans Candida prosthetic joint infections:A systematic review of treatment