
Extrahepatic hepcidin production: The intriguing outcomes of recent years
2019-08-14 07:31RaDaherThibaudLefebvreHervPuyZoubidaKarim
World Journal of Clinical Cases 2019年15期
Raêd Daher,Thibaud Lefebvre,Hervé Puy,Zoubida Karim
Abstract
Key words: Hepcidin; Extrahepatic hepcidin; Iron metabolism; Bacterial infection;Inflammation
INTRODUCTION
Iron is one of the most abundant metals in the planet with the potential of high toxicity to living cells.Highly active cells need iron for their metabolic activity because iron exhibits an optimal chemical property for electron transfer,facilitating biochemical reactions between different atoms and molecules.The toxicity of iron is due to induction of reactive oxygen species,which at high levels leads to cellular damage[1].
In the body,60% of iron is incorporated into hemoglobin,while 10% to 15% are found in muscle myoglobin and cytochromes.Circulating iron,related to transferrin,represents only 1%.Under physiological conditions,the liver and macrophages of the reticuloendothelial system are the main iron storage and recycling sites.One to two milligrams of iron are lost daily by sweating and desquamation of skin and intestinal cells,and in women by menstrual bleeding.This small amount is totally recovered by intestinal absorption of heme and non-heme iron,which takes place in the duodenal enterocytes[2].
There is no effective mechanism for iron excretion.As a result,the exogenous iron supplied to the body is not eliminated and may accumulate in a toxic way in the tissues.
BODY IRON METABOLISM
Dietary iron (Fe) III is reduced to FeII by duodenal cytochrome B reductase located in the brush borders of the enterocytes[3].FeII is then transported through divalent metal transporter 1 (named DMT1 or SLC11A2)[4]and is exported to bloodviaferroportin(FPN or SLC40A1),which shares no homology with DMT1 and is localized at the basolateral pole of the enterocytes[5].The export of iron by FPN requires a ferroxidase activity provided by the enzymes hephastin and/or ceruloplasmin allowing transferrin (Tf) to bind the circulating iron in the form of FeIII.Most cells in the body can assimilate Tf-bound iron through the ubiquitous transferrin receptor 1 (TfR1) that has a high affinity for the Tf-FeIII complex.After TfR1-mediated endocytosis,iron is released from Tf in the intracellular endosomes at acidic pH.The released FeIII is then reduced to FeII by a reductase,for example the six epithelial transmembrane antigen of the prostate 3 in erythroid cells.FeII is then transported to the cytosol through DMT1 localized in these endosomes.Erythroid cells of the bone marrow are the largest consumers of iron.About one billion iron atoms (20 to 30 mg) are used daily to form hemoglobin in newly produced erythrocytes.Macrophages of the spleen and liver recover heme iron from senescent erythrocytes after phagocytosis and catabolism of heme by the enzyme heme oxygenase[6].The mobilizable iron is thus recycled to the plasma for redistribution to tissues.FPN is essential for the export of iron by macrophages[7].
In all cells,unused iron is stored in a non-reactive form due to ferritin (Ft).FeII could be delivered to Ft by cytoplasmic chaperones,such as poly (rC)-binding protein 1[8].Ft is a protein complex consisting of 24 subunits of two types called heavy and light ferritins.Each complex is capable of storing up to 4500 iron atoms,which are easily mobilized when needed.A secreted form of Ft,low in iron,is found in the plasma.This serum Ft,used clinically to evaluate iron stores,is composed mainly of light subunits,some of which are glycosylated.In the majority of cases,serum Ft concentrations correlate with tissue stores of iron except under certain conditions where the synthesis of Ft is mainly due to inflammation.Indeed,Ft stimulated by inflammatory cytokines[9]is widely recognized as a marker of acute and chronic inflammation.
REGULATION OF IRON METABOLISM
Intracellular regulation
Intracellular iron homeostasis is ensured by the iron response element (IRE)-iron regulatory protein (IRP) system that controls the post-transcriptional expression of several iron proteins (TfR1,DMT1,FPN,Ft,and ALAS2; the first enzyme involved in heme biosynthesis in erythroid cells)[10-13].IREs are hairpin-like RNA motifs that serve as specific binding sites for IRPs.IRPs are soluble cytosolic proteins whose activity varies according to the intracellular iron concentration.There are two IRPs:IRP1 and IRP2 with a sequence identity of 56%[14].The IRE-IRP interaction stabilizes the mRNA when the IRE motifs are located in the 3'-UTR region,such as for TfR1,and imposes a steric constraint on the translation of the protein when IRE motifs are located in the 5'-UTR region.This results in an increase in transferrin-related iron acquisition when intracellular iron levels are low.A decrease in FPN and ferritin expression also occurs because iron export and intracellular iron storage are not appropriate in this condition.
Systemic regulation
Hepcidin was initially identified as an antimicrobial peptide[15](see section below).Yet since 2001,it is considered to be the master regulator of iron balance in humans by decreasing iron absorption and increasing iron retention in macrophages and Kupffer cells[16,17].The mechanism by which hepcidin inhibits these iron effluxes has been well studied in macrophages,where hepcidin binds to FPN and leads to its internalization and degradation in lysosomes[18,19].In duodenal cells,studies have reported a different mechanism.Indeed,hepcidin was shown to decrease duodenal transepithelial iron transport without internalization of FPN from the plasma membrane[20-22].In these cells,,hepcidin leads to a decrease in the protein expression of DMT1 rather than that of FPN[21].Recently,the non-internalization of FPN following hepcidin treatment was also shown in other cellular contexts[23,24].Zhanget al[23]first observed that FPN was highly expressed in mature red blood cells lacking the proteasomal degradation pathway.In these mature red blood cells,hepcidin was shown to inhibit iron export but did not change FPN abundance similarly to what we have previously observed in duodenal enterocytes[21].Studies on hepcidin-FPN interactions in the transfected HEK293T cell line andXenopusoocytes also confirmed that hepcidin binding is able to block iron export without causing FPN internalization[24].Thus,it seems that depending on the cellular and membrane environment hepcidin binding may block iron efflux acting either on FPN activity and/or on its abundance at the cell membrane.Moreover,in the duodenum,a permanent increase in hepcidin,as in a transgenic mouse model for example,has been shown to ultimately reduce FPN abundance by mechanisms that remain to be explored[25].
HEPCIDIN PRODUCTION
Hepcidin is synthesized in the liver as a pre-propeptide of 84 amino acids (aa); it then undergoes successive proteolytic cleavage to produce the 25-aa bioactive form,which is secreted in plasma.Being a low-molecular-weight peptide,hepcidin is assumed to be rapidly excreted by the kidney[26,27],where it is supposed to be taken up and degraded by the renal proximal tubule.Indeed,hepcidin was first isolated from human urine and named according to its synthetic site (hep-) and its antibacterial properties shownin vitro(-cidin).The mass spectroscopy studies have shown the presence of urinary 25-aa hepcidin as well as shorter forms (22 aa and 20 aa) that are supposed to be degradation products with still unknown functions[28].Hepcidin exhibits a cysteine-rich structure reminiscent of four disulfide bridges defensins within vitroantimicrobial activity[15,26,29],suggesting ancestral innate defense properties against invasive bacteria.Hepcidin structure is characteristic of peptides able to disrupt bacterial membranes and is similar to other antimicrobial peptides such as defensins.Hepcidin may also limit bacterial proliferation by decreasing iron in plasma and extracellular fluids.With regards to this immune activity,hepcidin synthesis was shown to be highly induced by inflammatory signals such as IL-6,allowing it to play a major role in the anemia associated with chronic diseases and inflammation[30-33].Hepcidin is also induced in the liver in response to lipopolysaccharide (LPS) through activin B and SMAD-signaling,but this pathway is still debatable[34-37].
Hepcidin is mainly,but not exclusively,produced and secreted by the liver.Indeed,several studies have shown that hepcidin is locally synthesized by multiple other tissues including kidney[38],macrophages[39,40],stomach[41],adipose tissue[42],brain[43],heart[44]and pancreas[45].Iron is essential for the normal function of these organs.In addition,hepcidin was found in atypical biological fluids as bile,ascitic and pleural,and cerebrospinal liquid[46,47].The functions carried out by this extrahepatic hepcidin are not completely known but this concern has gained increased attention in recent years.Some related findings will be discussed below.
Hepatic hepcidin
Serum levels of hepcidin are mostly correlated with the levels of liver hepcidin expression[48]demonstrating that hepatic hepcidin is the key regulator of systemic iron balance.This was obviously demonstrated in mouse models with either total or liverspecific ablation of the hepcidin gene[49,50].Liver-specific knockout (KO) mice were shown to fully recapitulate the severe iron overload phenotype observed in the total KO mice with increased plasma iron and massive parenchymal iron accumulation.
The regulation of hepatic synthesis of hepcidin is extremely complex and responds to multiple signals,some of which are still unclear.Indeed,the synthesis of hepcidin is stimulated by the high intake of iron and by inflammation,while it is repressed by iron deficiency and by all the pathological situations that stimulate the erythropoietic activity (anemia,bleeding,hemolysis,dyserythropoiesis and erythropoietin injections).
Hepatic hepcidin is regulated by iron-mediated pathways through a complex of integral hemochromatosis proteins.Hemojuvelin (HJV) was first identified as the mutated protein in the majority of juvenile hemochromatosis[51].The absence of HJV leads to a severe deficit in hepcidin production.HJV acts as a co-receptor with bone morphogenetic proteins (BMPs),enhancing activation of the SMAD pathway in response to binding of BMPs to their receptor (BMPR).BMP6 plays a major role in this pathway and in the stimulation of hepatic hepcidin by iron.In mice,the ablation of theBmp6gene causes severe iron overload[52].In humans,we and others[53-57]recently identified heterozygous mutations localized in the propeptide of the BMP6 protein leading to mild to moderate hemochromatosis.The BMP6 pathway is negatively controlled by matriptase 2 that interrupts the binding of BMP6 on its BMPR in hepatocytes[58-61].The BMP6 pathway also seems to be a target of erythroferrone,the erythroid factor that represses hepcidin when erythropoiesis is pathologically stimulated[62].
The study of the different forms of genetic hemochromatosis has demonstrated the role of the human hemochromatosis protein (HFE) and transferrin receptor 2 (TfR2) in the regulation of hepcidin by iron.The forms of adult hemochromatosis are due to mutations of theHFEgene for the most common forms,or TfR2 for rarer forms and are characterized by a lack of activation of hepcidin in response to iron overload[63].Mice deficient inHfeor patients withHFEmutations have a low hepcidin mRNA level in the liver despite their iron overload.A model has been proposed in which HFE,TfR2 and HJV interact with each other at the hepatocyte membrane to form an “ironsensing complex”[64].When transferrin saturation in serum increases,holo-transferrin shifts HFE from its binding to TfR1,which allows its interaction with TfR2 and activates transcription of hepcidin-encoding gene.Thus,TfR2 acts as a sensor of the transferrin saturation (serum iron) and BMP6 as a tissue iron sensor activating hepcidin synthesis by interaction with HJV during excessive accumulation of iron in hepatocytes[65,66].
Extrahepatic hepcidin
Hepcidin expression in the kidney: In 2005,using immunocytochemistry assays,Kulaksizet al[38]observed for the first time that hepcidin was expressed in the kidney,namely in the cortical thick ascending limb (cTAL) and connecting tubules and to a lesser extent in the collecting ducts.Hepcidin was absent in the proximal tubule and descending and ascending thin limbs[38].It was localized at the apical surface of the renal epithelial cells,which suggests that renal hepcidin is eliminated in the urine after an autocrine/paracrine action on renal tubules.Using microdissected and isolated renal tubules,we also confirmed that hepcidin is preferentially expressed throughout the distal nephron,particularly in the TAL[67].Interestingly,the distal nephron expresses DMT1 and FPN transporters at the apical and the basolateral membrane,respectively and was described to be the site of the reabsorption of nonheme iron in kidney[68-71].Using mouse models with defects in hepcidin production(hepcidin KO and HJV KO mice),our group had clearly shown that non-heme iron accumulated in this distal nephron particularly the TAL[71].In the TAL cell line,we found that vectorial transport of iron was decreased following exogenous hepcidin treatment.In addition,similarly to what we observed in intestine[21],hepcidin was able to decrease DMT1 bothin vivousing kidney sections from hepc-/- mice andin vitrousing TAL cells.All together,these reports highlight a new role of hepcidin in the control of renal iron transport and accumulation and suggest that local synthesis of hepcidin within renal tubules may play a crucial role in this effect.Hepcidin seems to specifically target the distal nephron rather than the proximal tubule.We investigated the importance of hepcidin in the protection of kidneys against urinary tract infection[67].We developed an experimental urinary tract infection model and showed that hepcidin KO mice inoculated with the gram-negative CFT073 strain exhibited higher renal bacterial load than infected wildtype mice as well as a significant attenuation of renal inflammatory profile.Hepcidin KO mice showed a marked alkalization of urine associated with repression of both Atp4a and Atp12a proton pumps.Pre-treatment of wildtype mice with hepcidin considerably reduced renal colonization by the CFT073 strain and restored the acidic pH of urine.In vitroexperiments proved the bacteriostatic activity of hepcidin against UroPathogenicEscherichia colibecause the bacterial growth was inhibited in the presence of this peptide.Interestingly,we found that CFT073 repressed renal hepcidin expression in infected mice through reduction of SMAD signaling.These results indicate that hepcidin plays a role in the fight against UroPathogenicEscherichia coliinfection and that UroPathogenicEscherichia colimight target renal hepcidin to attenuate its global antibacterial activity in the early phase of a UTI[67].
Hepcidin expression in macrophages: Macrophages play a central role in iron recycling and consequently in establishing iron balance.Initial studies from Liuet al[39]demonstrated that the reticuloendothelial system (RES,namely liver Kupffer and spleen macrophages) was able to produce hepcidin following LPS-induced inflammation in mice and upon treatment of mouse splenic adherent cells by LPS.This RES-produced hepcidin was independent of the iron pool of the cells becausein vivoiron loading has been shown to induce hepcidin production in liver without increase in hepcidin mRNA in the spleen.In addition,when splenic cells were treatedin vitrowith ferric ammonium citrate,there was again no increase in splenic hepcidin mRNA expression.Peyssonnauxet al[40]also investigated the ability of macrophages to endogenously synthesize hepcidinin vitroandin vivofollowing bacterial infections.They found that intraperitoneal bacterial challenge was able to increase hepcidin expression in splenic macrophagesviathe LPS-mediated Toll-like receptor 4 pathway.In addition to RES,hepcidin was shown to be produced by alveolar macrophages at a low level when not stimulated but at a high level when exposed to LPSin vitroandin vivo[72,73].In agreement with these previous data,Sowet al[74]also showed a strong induction of hepcidin mRNA expression in several macrophage cell lines infected with mycobacteria and/or treated with the inflammatory cytokine IFN-γ.
The physiological significance of local hepcidin expression in macrophages is not yet fully determined but the hypothesis is that RES-produced hepcidin may contribute by greatly increasing the regulatory pool of hepcidin around liver Kupffer cells and spleen macrophages as well as other macrophages residing in tissues frequently confronted by infection.Increased local hepcidin might potentiate the retention of iron during conditions of inflammation and infection particularly since FPN is strongly expressed at the cell surface of these cells.These findings strongly suggested that macrophage hepcidin could play a role in host defense by acting on FPN expression and consequently limiting the availability of iron to invading bacteria.
Recently,we described the involvement of RES-produced hepcidin in iron metabolism dysregulation in Gaucher disease (GD),an inherited deficiency of the lysosomal enzyme glucocerebrosidase leading to accumulation of glucosylceramide in tissues including the spleen and the liver[75].In order to understand the unexplained hyperferritinemia frequently reported in Gaucher disease patients,Perl’s staining of spleen and bone marrow smears was performed.Analysis revealed iron accumulation in the lipid-laden macrophages also called Gaucher cells.Using anin vitromodel of Gaucher cells,we have shown that hepcidin production was induced in these macrophages,and FPN protein was consequently internalized from the plasma membrane.Thus,hyperferritinemia in Gaucher disease could be related to the sequestration of iron in Gaucher cells due to local production and an autocrine action of hepcidin in these cells.
Hepcidin expression in the stomach: The gut is an additional organ that produces hepcidin[41].In the stomach,hepcidin was found in the gastric fundus and corpus,more precisely,in parietal cells.In vitro,gastric hepcidin was upregulated upon treatment with IL6 and in response to bacterial infection.In humans,gastric hepcidin level was elevated during bacterial infection and then normalized after successful eradication.In addition,acid secretion in hepcidin KO mice was markedly reduced due to repression of Atp4a proton pump,and this was associated with gastric bacterial overgrowth.Thus,this original study was the first to show that hepcidin is a gastric factor essential for acid secretion and fight againstHelicobacterbacterial invasion.
Hepcidin expression in adipose tissue:Hepcidin synthesis in adipose tissue was reported by Bekriet al[42]in severely obese patients with low-grade systemic inflammatory disorder[42].mRNA level of adipose tissue hepcidin was correlated with multiple indexes of inflammation (IL6,C-reactive protein).This observation was also confirmed byin vitrostudies using cultured adipose tissue explants,where IL6 was shown to promote hepcidin expression.Thus,adipose tissue hepcidin is suggested to exacerbate the iron deficiency and iron deficiency anemia observed in the majority of obese patients.In this context,it is not clear whether adipose tissue hepcidin contributes to increased pool of systemic hepcidin,but one can suppose that adipose tissue may provide a high concentration of local hepcidin around intestinal cells and inhibit iron absorptionviaan autocrine/paracrine mechanism.Yet,numerous studies have been conducted to explore the role of hepcidin secreted by adipose tissue in obesity hypoferremia.Several high-fat diet mouse models were developed for that purpose,but the results remained highly controversial[76-80].
Hepcidin expression in the brain:Iron has a role in oxidative metabolism and is a cofactor in the synthesis of myelin and neurotransmitters.It is thus essential for normal neurological function.The presence of hepcidin in the brain was shown by Zechelet al[43].Using different RNA and protein detection techniques,the cellular distribution of hepcidin in murine brain was investigated.Results showed a widespread distribution of this peptide in different brain areas.Many other reports described the presence of brain hepcidin production although to a lesser extent with protein levels higher than mRNA levels,suggesting that the liver is partially responsible in addition to locally produced hepcidin[81,82].
Hepcidin expression in the brain is induced by inflammation.LPS and IL6 are known as major actors in inflammation-induced mechanisms.LPS was described as an indirect inductor of hepcidin expression in astrocytesviaupregulated IL6 expression in microglia[83,84].However,other experiments showed a direct upregulation of hepcidin by LPS in glial cells[85].
Recently,we investigated iron metabolism in the brain of a mouse model of Sanfilippo syndrome (mucopolysaccharidosis type III) where the progressive accumulation of heparan sulfate oligosaccharides induced microglia and astrocytes to produce pro-inflammatory cytokines leading to severe neuroinflammation[86].We found that iron accumulation in mucopolysaccharidosis type III mice mainly affected the cerebral cortex where hepcidin expression was higher than in wildtype mice.This increase was correlated with low expression of FPN and consequently brain iron retention.We showedin vitrothat heparan sulfate oligosaccharides are directly responsible for the induction of hepcidin and a decrease in FPN level when added to microglia and to a lesser extent to astrocytes.Our results showed that microglia play a key role in brain hepcidin overexpression,and the regulation of brain hepcidin may be dependent on or independent of inflammation.
Because hepcidin may act as an antimicrobial peptide in the brain,we investigated hepcidin production in cerebrospinal fluid during infectious meningitis.Using a liquid chromatography tandem mass spectrometry method[48],we compared hepcidin signal in cerebrospinal fluid of 5 patients diagnosed for viral meningitisversus5 patients with bacterial meningitis.In viral infection,hepcidin signal was very low nearing the background level in the same range as healthy patients (Figure 1).By contrast,cerebrospinal fluid hepcidin was significantly increased in bacterial meningitis.These observations were different from what was observed in the liver where hepcidin was induced by both bacterial and viral infections,suggesting again that hepcidin plays tissue-specific roles[87].The most frequent pathogens in community-acquired bacterial meningitis are pneumococcus and meningococcus,which are gram-negative germs and LPS producers[88].Thus,like what was described in the kidney[67]and elsewhere[89],brain hepcidin must be more sensitive to gramnegative bacteria and induced through the LPS/Toll-like receptor signal transduction pathway.
Hepcidin expression in the heart:Iron is essential for normal heart function;however,the dysregulation of cardiac iron homeostasis may be deleterious.Merleet al[44]performed the first analysis of hepcidin expression and its regulation in rat heart.They reported that hepcidin is expressed in cardiomyocytes,and it is regulated in response to hypoxia and inflammation,which strongly suggests that this peptide may play an important role in cardiac diseases.Quantification of hepcidin postulated the heart as the organ with the highest hepcidin level next to the liver[15,90].Geet al[91]studied the effect of hepcidin on FPN expression in cardiomyocytes.Using a cardiomyocyte cell line,they demonstrated that local hepcidin was able to reduce FPN level and iron export from these cells.Studies conducted by Lakhal-Littletonet al[92]strongly supported these data.Indeed,they first confirmed that FPN is expressed in cardiomyocytes and demonstrated that its cardiac-specific deletion leads to fatal cardiac iron overload.To go further,they generated mice with cardiomyocyte-specific hepcidin deletion or knock-in of hepcidin-resistant FPN.They found that both models maintained normal systemic iron homeostasis but developed fatal cardiac dysfunction as a consequence of cardiomyocyte iron deficiency[93].Thus,they provided evidence for a cell-autonomous role of hepcidin in cardiac iron homeostasis.
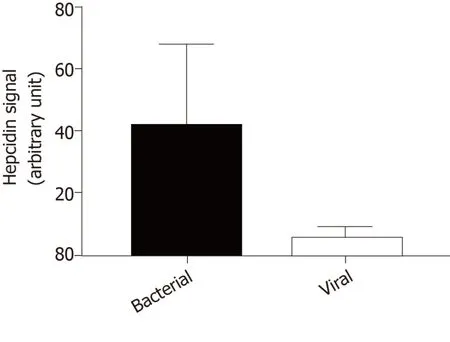
Figure 1 Hepcidin signals measured by liquid chromatography tandem mass spectrometry in cerebrospinal fluid of patients with bacterial meningitis and viral meningitis (n = 5 per group).
Hepcidin expression in the pancreas:Data published by Kulaksizet al[45]demonstrated that hepcidin is expressed in the pancreas of rat and human.Further analysis showed that it was localized in β-cells of the islets of Langerhans.In addition,thein vitroexperiments performed in this study demonstrated that the expression of hepcidin in β-cells is directly regulated by iron.
Iron is important for normal insulin secretion.However,excess of iron have been shown to affect β-cell function in hemochromatosis models[94-96],causing iron accumulation in the islets,decreased insulin secretion and increased apoptosis.In contrast,iron pool decrease was shown to protect from diabetes and loss of β-cell function in the obese (ob/ob) mouse model[97].Both DMT1 and FPN are expressed in β-cells.In β-cell-specific DMT1 KO islets,glucose-stimulated insulin secretion was reduced[98].These observations suggested that hepcidin produced by β-cells may be involved in an intrinsic regulation of pancreas iron and in their function in glucose homeostasis.
CONCLUSION
Although only some organs have been addressed in this review,there is a number of studies describing the production of hepcidin in many others such as lungs[99,100],prostate[101],placenta[102,103]and retina[104].Our hypothesis is that peripheral hepcidin is intended for an innate immune response including a defense against bacterial invasion.However,under local or systemic inflammatory conditions,the induction of this peripheral hepcidin may contribute to target tissue damage due to local accumulation of toxic iron and apoptosis.Nevertheless,despite considerable advances recently,further explorations deserve to be rapidly achieved to deeply investigate the cellular mechanisms and functions of peripheral hepcidin as well as its regulation in the different organs.
 World Journal of Clinical Cases2019年15期
World Journal of Clinical Cases2019年15期
- World Journal of Clinical Cases的其它文章
- Bone alterations in inflammatory bowel diseases
- Neoadjuvant endocrine therapy: A potential strategy for ER-positive breast cancer
- Vestigial like family member 3 is a novel prognostic biomarker for gastric cancer
- HER2 heterogeneity is a poor prognosticator for HER2-positive gastric cancer
- Changes in corneal endothelial cell density in patients with primary open-angle glaucoma
- Myocardial bridge-related coronary heart disease: lndependent influencing factors and their predicting value