
Acute kidney injury spectrum in patients with chronic liver disease:Where do we stand?
2019-08-12 02:45:08WiwatChancharoenthanaAsadaLeelahavanichkul
World Journal of Gastroenterology 2019年28期
Wiwat Chancharoenthana, Asada Leelahavanichkul
Abstract Acute kidney injury (AKI) is a common complication of liver cirrhosis and is of the utmost clinical and prognostic relevance. Patients with cirrhosis, especially decompensated cirrhosis, are more prone to develop AKI than those without cirrhosis. The hepatorenal syndrome type of AKI (HRS-AKI), a spectrum of disorders in prerenal chronic liver disease, and acute tubular necrosis (ATN) are the two most common causes of AKI in patients with chronic liver disease and cirrhosis. Differentiating these conditions is essential due to the differences in treatment. Prerenal AKI, a more benign disorder, responds well to plasma volume expansion, while ATN requires more specific renal support and is associated with substantial mortality. HRS-AKI is a facet of these two conditions,which are characterized by a dysregulation of the immune response. Recently,there has been progress in better defining this clinical entity, and studies have begun to address optimal care. The present review synopsizes the current diagnostic criteria, pathophysiology, and treatment modalities of HRS-AKI and as well as AKI in other chronic liver diseases (non-HRS-AKI) so that early recognition of HRS-AKI and the appropriate management can be established.
Key words: Acute kidney injury; Acute-on-chronic liver failure; Chronic liver disease;Hepatorenal syndrome; Plasma perfusion and bilirubin adsorption and double plasma molecular absorption system; Fractionated plasma separation and adsorption; Molecular adsorbent recycling system; Single-pass albumin dialysis
INTRODUCTION
Acute kidney injury (AKI) superimposed on chronic liver disease and cirrhosis is common and consists of varying phenotypes. Prerenal renal dysfunction caused by severe hypoalbuminemia is the most common clinical syndrome in patients with advanced liver disease. Although prerenal azotemia seems to be the first phase of AKI, it is difficult to differentiate from hepatorenal syndrome (HRS) and acute tubular necrosis (ATN). Once the onset of AKI occurs in chronic liver disease, a consequence of its complications is an increased morbidity and mortality rate. Accordingly, the characteristics of renal dysfunctions in both noncirrhotic and chronic liver diseases,such as prerenal HRS-ATN, require not only earlier recognition but also precise diagnosis with optimal management. Recently, there have been great advances,including in classification, nomenclature, and pathophysiology, in identifying the connection between chronic liver disease and acute renal dysfunction in patients with cirrhosis. Indeed, acute-on-chronic liver failure (ACLF) was first recognized in the early 2000s as a new classification that represents the distinct characteristics of chronic liver failure (or decompensated cirrhosis) with rapid deterioration leading to hepatic and extrahepatic multiorgan failure[1,2]. Since then, many efforts in natural history and pathophysiology have been made[3]. Substantial advancements have been made not only in the field of hepatology but also in the understanding of the significance of renal dysfunction in chronic liver disease, as suggested by the international guideline that all acute renal dysfunction in patients with cirrhosis requires the same clinician attention as AKI[4].
Since the first description by Hecker and Sherlock in the 1960s, renal dysfunction in patients with ascites and advanced cirrhosis has typically been called HRS[2], which refers to a syndrome of decreased renal function mainly resulting from the systemic hemodynamic effects of advanced portal hypertension. However, advanced chronic liver disease can impact renal function with a wide range of complications, including bile acid nephropathy, coagulopathy-induced bleeding from ischemic ATN, related glomerular diseases (e.g., immunoglobulin, a nephropathy, hepatitis B-related glomerulonephritis, hepatitis C-related glomerulonephritis, cryoglobulinemia,membranoproliferative glomerulonephritis), and other comorbid diseases such as inherited cystic diseases. Herein, we review the updated information, including the etiology, pathophysiology, and therapeutic aspects, of renal dysfunction in advanced chronic liver disease.
NOMENCLATURE AND CLASSIFICATION OF ADVANCED LIVER DISEASES
The clinical spectrum of advanced liver disease is currently recognized as follows:
Chronic liver disease
Chronic liver disease is a progressive process of destruction and regeneration of liver parenchyma resulting in fibrosis and cirrhosis over a period of 24 wk (Figure 1)caused by inflammation, infection, abnormal metabolism, or malignancy. The functional classification of chronic liver disease can be divided into compensated and decompensated liver disease.
Acute-on-chronic liver failureACLF is liver failure with one or more extrahepatic organ failures, leading to an increased 28-d mortality rate within 3 mo of disease onset[5]. However, there are currently at least 4 different definitions for ACLF in clinical practice: (1) The Asian Pacific Association for the Study of Liver (APASL); (2) The American Association for the Study of Liver Diseases (AASLD); (3) The European Association for the Study of the Liver (EASL) or AASLD-EASL; and (4) The World Gastroenterology Organization(WGO) definition (Table 1).
The first ACLF definition, given by the APASL in 2009, was “acute hepatic insult manifesting as jaundice, and coagulopathy complicated within 4 wk by ascites and/or encephalopathy in a patient with previously diagnosed or undiagnosed chronic liver disease”[6]. Then, in 2014, the revised ACLF definition included the presentation of a high mortality rate within 28 d of disease onset (short-term mortality)[7]. Later, the AASLD-EASL workgroup defined ACLF as “acute deterioration of preexisting chronic liver disease usually related to a precipitating event and associated with increased mortality at 3 mo due to multisystem organ failure”, based on the data from large prospective multicenter studies[8,9]. After that, the WGO workgroup announced an improved ACLF definition: “a syndrome in patients with chronic liver disease with or without previously diagnosed cirrhosis which is characterized by acute hepatic decompensation resulting in liver failure (jaundice and prolonged international normalized ratio) and one or more extrahepatic organ failure that is associated with increased mortality within a period of 28 d and up to 3 mo from onset”[5].Interestingly, while the timeframe between the onset of acute liver injury and liver failure development (4 wk) is clear in the APASL definition, time intervals are not mentioned in the AASLD-EASL or the WGO definition.
In addition, there are some differences in ACLF definition regarding the underlying liver conditions of the patients. Patients with cirrhosis and non-cirrhosis (i.e., precirrhotic) are included in the APASL and WGO but not in the AASLD-EASL definition. Patients with hepatic decompensation (previously or currently) are not included in the APASL definition, while they are included in the AASLD-EASL definition (which does not include noncirrhotic chronic liver disease). In parallel, the WGO definition includes both cirrhosis (compensated and decompensated) and noncirrhotic chronic liver disease.
Because ACLF can develop from: (1) Noncirrhotic liver disease (e.g., hepatitis B,alcohol-related chronic liver disease, and nonalcoholic fatty liver disease (NAFLD); (2)Compensated cirrhosis with precipitating factors; and (3) Decompensated cirrhosis,the WGO suggested that all patients with chronic liver disease (no cirrhosis,compensated cirrhosis, or decompensated cirrhosis) should be included in the ACLF definition and divided into types A (noncirrhotic chronic kidney disease), B(compensated cirrhosis), and C (decompensated cirrhosis)[5](Figure 1).
THE PARADIGM SHIFTS IN THE DEFINITION AND PATHOPHYSIOLOGY OF RENAL DYSFUNCTION IN LIVER DISEASE
The diagnostic criteria of AKI in patients with cirrhosis: an updated definitionHistorically, AKI in patients with cirrhosis was often defined as HRS type 1 and type 2. HRS type 1 referred to AKI in cirrhosis with a relatively rapid progressive deterioration of renal function with serum creatinine (SCr) higher than 2.5 mg/dL for more than 2 wk, and HRS type 2 referred to AKI with slowly progressing renal function deterioration in cirrhosis with refractory ascites (SCr level of 1.5-2.5 mg/dL)[10]. Because SCr is a less sensitive biomarker of AKI, with several pathophysiological limitations due to sarcopenia caused by hepatic injury (discussed later), the International Club of Ascites (ICA) recently proposed a new diagnostic criterion for HRS in 2015[4]. Accordingly, the SCr > 2.5 mg/dL criterion was removed along with recommending HRS as another kind of AKI, called HRS-AKI. In addition,the 2-wk threshold for the diagnosis of HRS and its subtypes was removed. The ultimate changes to the diagnostic criteria from HRS to HRS-AKI are now in the context of “cirrhotic patients who develop AKI by detecting a change in absolute SCr level of ≥ 0.3 mg/dL or by an increase in SCr ≥ 50% from baseline within 48 h with a lack of volume expansion response without evidence of shock, recent exposure to nephrotoxic agents or preexisting structural renal disease”[4](Table 2).

Figure 1 Definition of liver disease terminology according to timing and characteristics. ACLF: Acute-on-chronic liver failure; AKI: Acute kidney injury; HRS:Hepatorenal syndrome.
In 2007, the Acute Kidney Injury Network (AKIN) proposed the guidelines for AKI-defining criteria[11], which were integrated into the ICA and the Acute Dialysis Quality Initiative (ADQI) in 2011[12](Table 3), because several validation studies on the AKIN criteria show the independent mortality association depending on the stage of renal injury[13-16]. The most recently proposed guideline from Huelin et al[17]suggests that stage 1 AKI should be divided into 2 phases, stage 1A (SCr < 1.5 mg/dL) and stage 1B (SCr > 1.5 mg/dL), because stage 1 AKI with SCr > 1.5 mg/dL shows a similar mortality rate as stage 2 AKI. However, further validation study is needed due to the unclear differences in baseline characteristics between stage 1A and stage 1B AKI, where the more severe liver dysfunction is shown in the latter group.
Hepatorenal syndrome-acute kidney injury: A changing of pathophysiologic definitionsThe new definitions of advanced liver disease and AKI improve the understanding of the underlying pathophysiologic mechanisms in the liver and kidneys. HRS is currently not the only form of AKI in chronic liver disease. Indeed, a vast majority of AKI in chronic liver disease is not characterized as HRS, particularly in ACLF (Table 4).
Splanchnic vasodilatationThe progression of advanced chronic liver disease into liver cirrhosis (liver architecture disruption) results from splanchnic vasodilatation in response to increased intrahepatic resistance (portal hypertension) caused by several mediators,including nitric oxides, prostacyclin, carbon monoxide, epoxyeicosatrienoic acids,glucagon, endogenous cannabinoids, and adrenomedullin[18-24]. In advanced cirrhosis,the loss of liver compensation that slows disease progression in the liver is due to progressive splanchnic vasodilatation (loss of the counteract vasoconstriction),leading to a decrease in the effective circulatory volume, reduced renal blood flow and AKI. This phenomenon simulates the renin-angiotensin-aldosterone system(RAAS) and vasopressin, which in turn cause more severe renal vasoconstriction and worsening renal hypoperfusion[25]. Evidence of their functional nature was illustrated by measuring copeptin, a 39-amino-acid glycopeptide released from the neurohypophysis, and arginine vasopressin (AVP). The higher copeptin level is, the greater the risk of AKI and the worse the outcomes of decompensated cirrhosis[26]. In addition, the deterioration of renal perfusion is caused by the alteration of renal blood flow autoregulation[27,28]. Together, the current treatment concepts of HRS-AKI are reversing the physiological responses by improving the hemodynamic vascular bed using volume expansion (such as with albumin) together with splanchnic vaso-constrictors.
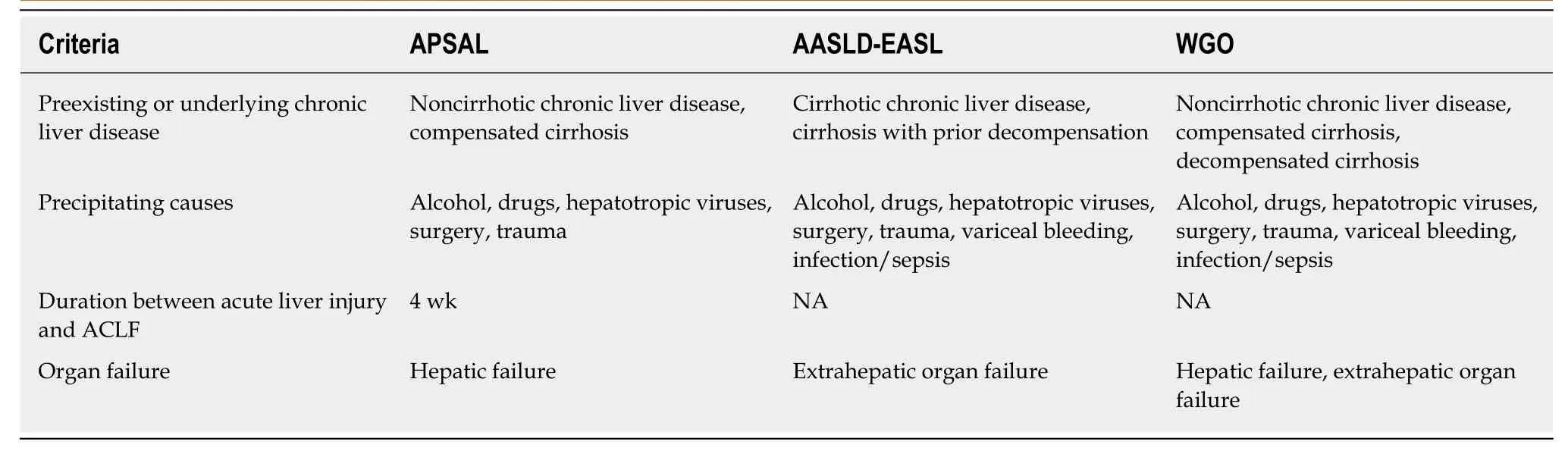
Table 1 Comparison of the definitions of acute-on-chronic liver failure from the Asian Pacific Association for the Study of Liver,American Association for the Study of Liver Diseases-European Association for the Study of the Liver, and World Gastroenterology Organization
Role of inflammationSystemic inflammation is one of the mechanisms responsible for decompensated cirrhosis because increased inflammatory cytokines are demonstrated in HRS-AKI(the late stage of cirrhosis). Advanced liver disease with spontaneous bacterial peritonitis (SBP) and renal insufficiency is characterized by high tumor necrosis factor(TNF)-α and interleukin (IL)-6 compared with advanced liver disease with normal renal function[29]. Furthermore, patients with acute decompensated cirrhosis and difficult-to-treat HRS-AKI (persistent AKI) demonstrate higher interferon-inducible protein-10 and vascular cell adhesion molecule-1 compared to patients with treatment-responsive HRS-AKI[30]. In addition, the growing evidence afforded by systems biology analysis has demonstrated a similar nature of inflammation in HRSAKI in comparison with AKI in chronic non-hepatic conditions, such as lupus nephritis[30].
Adrenal insufficiencyRelative adrenal insufficiency is found in over 25%-30% of decompensated cirrhosis patients. Of note, adrenal insufficiency contributes to cardiomyopathy in cirrhosis patients via the downregulation of β-adrenergic receptors as well as the alteration of catecholamines’ effects on the systemic vascular tone[31]. Furthermore, the presence of adrenal insufficiency in patients with stable decompensated cirrhosis is associated with poorer clinical characteristics, such as circulatory dysfunction, previous history of SBP, and worse survival rate[32].
Cardiac dysfunctionThe impacts of advanced chronic liver disease on cardiac function and the systemic circulatory system are well known. Cirrhotic cardiomyopathy is a diminished cardiac contractile function with electrophysiological abnormalities in response to stress stimuli without preexisting cardiac disease[33]. The emerging data reflect a liver-heartkidney interaction. Cirrhotic cardiomyopathy arises from sustained portal hypertension, which increases the risk of bacterial translocation and portosystemic shunt. Bacterial translocation stimulates the production of systemic inflammatory cytokines and eventually causes endothelial dysfunction[34], while an increased portosystemic shunt decreases systemic vascular resistance by increasing nitric oxide,adenosine, bradykinin, and endocannabinoids[35]. In addition, portal hypertension and splanchnic dilation activate the sympathetic nervous system and several neurohormones, including AVP, renin, and angiotensin, which affect heart function by increasing afterload and left ventricular end-diastolic pressure[36]. Decreased cardiac output and increased right atrial pressure cause renal tissue hypoxia and venous congestion, respectively, leading to kidney injury and lower glomerular filtration rate[37]. Accordingly, cardiac dysfunction in advanced chronic liver disease deteriorates renal function in HRS through a liver-heart-kidney connection, which is possibly related to a new model of therapeutic approaches to HRS[38].
Pathophysiology involved in AKI in other chronic liver diseases (non-HRS-AKI): An update of evidences

Table 2 The lnternational Club of Ascites diagnostic criteria for hepatorenal syndrome
Role of inflammation, apoptosis, and cell death: Hepatic inflammation is a novel major component for the initiation and progression of liver injury[39]. In chronic liver disease, hepatic inflammation is activated by several risk factors (i.e., systemic infection, gastrointestinal bleeding, alcohol, viral infection, etc.) resulting in (Figure 1):(1) Hepatocyte damage that delivering several damage-associated molecular patterns(DAMPs) from liver cells; and (2) Gut immunity impairment that enhancing the translocation of pathogen-associated molecular pattern (PAMPs) from gut organisms[40]. Subsequently, both DAMPs and PAMPs enhance the more severe liver damage (ALF, ACLF, and liver cirrhosis). Several liver associated DAMPs including IL-1, IL-33, high-mobility group box-1, and bile acid[41]are recognized by several receptors [e.g., toll-like receptors (TLR)-4, TLR-9 and the receptor for advanced glycation end-products] on Kupffer cells (liver macrophages)[42]resulting in the more enhanced hepatic damages (Figure 2).
In addition, the imbalance of inflammatory response in ACLF (a local inflammatory response) contributes to the systemic inflammatory response syndrome (SIRS) in different severity and subsequently turns into the compensatory anti-inflammatory response syndrome (CARS)[43]. While SIRS relates to the excessive liver inflammation and extrahepatic organ dysfunction, CARS is a counter regulatory mechanism against the inappropriate hyper-inflammatory process. Overwhelming of activated monocyte function may contribute to AKI, compatible with those in septic AKI[44]. In addition,Because macrophage polarization is related to pro- versus anti- inflammation, SIRS and CARS in ACLF might be associated with M1 and M2 macrophage polarization,respectively, of Kupffer cells in liver[45]. Moreover, bone marrow-derived macrophage that was recruited into liver by the inflammatory-induced CCL2 and CCR5 chemokine expression might also be important in liver injury[45]. Macrophage polarization and macrophage origin may be beneficial to the understanding in the inflammatory phase of liver injury (i.e., initiation, propagation and resolution)[46]. The inhibition of Kupffer cells, the prevention of monocyte recruitment into liver, and the promotion of proper macrophage polarization might be the novel strategies for liver injury attenuation[47].
Systemic oxidative stress and several inflammatory cytokines (human nonmercaptalbumin 2, IL-6, and IL-8) are also higher in patients with ACLF compared to non-ACLF conditions[48]. Similarly, the severity of ACLF is associated with apoptosis as determined by the apoptosis index [plasma caspase-cleaved keratin 18 (cK18; an apoptosis biomarker)/keratin 18 (K18; an indicator of total cell death)][49], implying the influence of systemic inflammation and apoptosis in ACLF. Indeed, persistent infection (inflammatory activation) is an important risk factor in cirrhotic patients on the basis of the score of chronic liver failure-organ failure. Therefore, AKI superimposed in ACLF is associated with a 20% increase in mortality depending on AKI severity (more severe with SCr > 1.5 mg/dL)[9].
Role of bile acid:The limited responsiveness of terlipressin plus albumin treatment in HRS type 1 with high serum bilirubin (≥ 10 mg/dL) suggests the influence of bile acid in chronic liver disease[50]. Indeed, the markedly elevated serum bilirubin in chronic liver disease induces bile cast nephropathy, a common renal pathology of AKI with severe liver dysfunction diagnosed from the presentation of intratubular bile casts by Hall histochemical staining, which are associated with irreversible AKI in chronic liver disease[51-53]. In the less severe form of renal injury, bile acid accumulation in cirrhosis induces proximal tubulopathy mimicking Fanconi syndrome (low uric acid,low phosphate but high bile acid in serum) without bile cast nephropathy[51-53].Although serum bilirubin is an independent predictor of the therapeutic response of HRS, the precise role of bile cast nephropathy is still unclear.

Table 3 The proposed classification system of renal dysfunction in patients with cirrhosis proposed by the Acute Dialysis Quality lnitiative and the lnternational Club of Ascites work group[12]
Worsening portal hypertension:Markedly increased intrahepatic resistance is common in progressive ACLF patients and usually results in increased portal hypertension[54]. This, in turn, may potentiate hepatorenal reflex disturbance, causing progressive AKI[27].
Worsening cardiac output:ACLF worsens cardiac functions through arterial vasodilation in the splanchnic area and peripheral circulation, which can be demonstrated in all stages of liver injury, from the early compensated stage to progressive liver decompensation to the late stage of HRS. The low cardiac output due to excessive vascular dilatation is counteracted by: (1) Several vasoconstriction mediators (including the RAAS, vasopressin, and the sympathetic nervous system);and (2) Vigorous salt and water balancing from renal homeostasis[55]. Once cardiac function fails to compensate for arterial vasodilation, as determined by the low cardiac output and reduced systolic function, the more severe liver injury condition(late HRS) will develop due to the decreased effective circulating volume[56].
TREATMENT
General managementAs discussed above, the points on the continuum of AKI disease in liver cirrhosis, i.e.,prerenal azotemia-HRS-ATN (prerenal-HRS-ATN), are difficult to differentiate clearly. AKI management in a certain setting should focus on early recognition with an understanding of the individual patient’s clinical course (Figure 3). The disturbance of hemodynamics in cirrhosis patients is the principal cause of AKI that is precipitated by infection, abrupt onset of severe hyperbilirubinemia, gastrointestinal bleeding, over-diuresis, or nephrotoxic agents. All of these factors must be immediately identified and corrected in patients with advanced cirrhosis to prevent subsequent renal complications. An intravascular volume status assessment is an initial step for any AKI etiologies. Volume status assessment is challenging,particularly in cirrhosis, because the patients usually also have hyperdynamic circulation between a total-body hypervolemic state and a low effective circulatory volume status. Unfortunately, there is no effective monitoring tool for these patients.Central venous pressure monitoring not only has a poor correlation with the intravascular volume due to confounding effects from ascites but also is too invasive for the coagulopathy that is commonly found in chronic liver disease.Echocardiography for intravascular volume assessment depends solely on the determination of inferior vena cava size and variability, which relies mostly on operator expertise[57].
Although there is no advantage in mortality attenuation or AKI incidence whencomparing albumin and crystalloids[58], albumin is usually the recommended volume expander in chronic liver disease due to albumin’s pleiotropic effects (antiinflammatory and antioxidant properties)[59,60]. Hydroxyethyl starch is contraindicated due to its associations with increased AKI and mortality rates[61]. In addition,intravenous albumin administration at 1-1.5 and 1 g/kg on days 1 and 3, respectively,with antibiotics attenuates the mortality of SBP and the AKI incidence[62]. In parallel,SBP prophylaxis with oral quinolones (norfloxacin 400 mg twice a day for 7 d) is recommended in the high-risk group (low protein in ascites fluid and previous history of SBP)[63], while intravenous ceftriaxone (1 g/d for 7 d) is more proper than oral quinolones in patients with active gastrointestinal bleeding[64]. Further, patients with high serum bilirubin (> 20 mg/dL) (with an abrupt onset or long duration) may develop nephropathy due to jaundice or bile cast induction[52,65], and selective bilirubin removal might be beneficial[66].
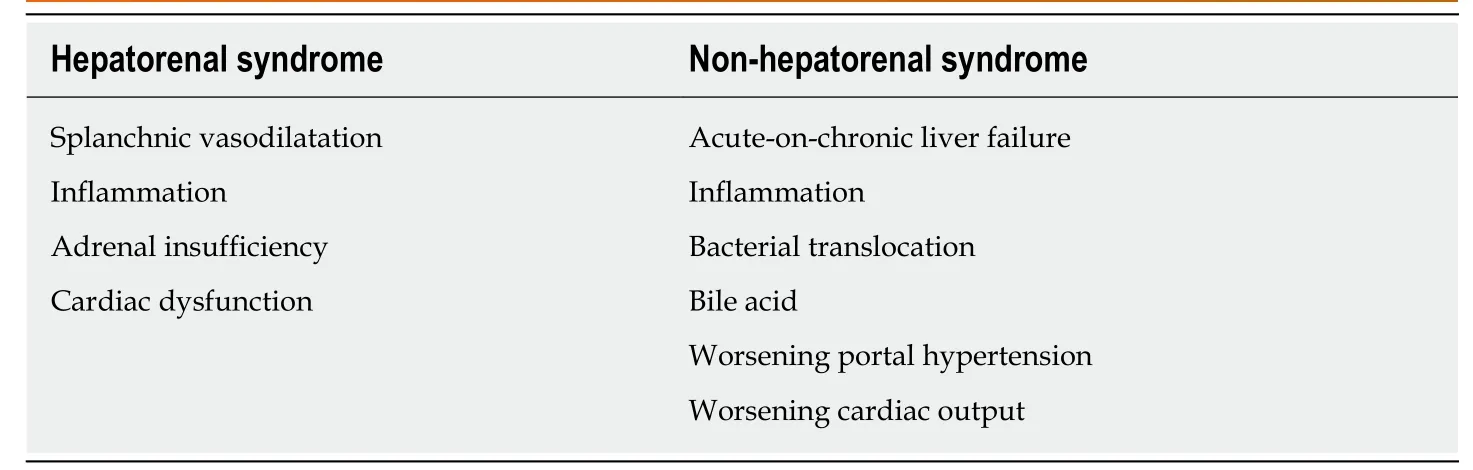
Table 4 Comparison between the main mechanisms of the pathophysiology of hepatorenal syndrome-acute kidney injury and non-hepatorenal syndrome-acute kidney injury
Intravascular volume depletion due to excessive diuretic treatment (diureticinduced AKI) is common. Furthermore, electrolyte disturbance, such as hyperkalemia from aldosterone antagonists or other potassium-sparing diuretics, may necessitate urgent renal replacement therapy, particularly in those with renal impairment.Hypokalemia, a precipitating factor of hepatic encephalopathy, is a frequent diuretic complication. Accordingly, diuretic administration should be reserved for patients with a hypervolemic state or marked ascites and should be avoided in those vulnerable to intravascular volume depletion. All nephrotoxic agents, such as radiological contrasts, need to be prescribed with caution. Sepsis is also a common condition that is found with decompensated cirrhosis[67]. Organ failures such as AKI and acute respiratory distress syndrome may be a result of a high production of sepsis-induced proinflammatory cytokine production[67]. Cirrhosis patients with AKI should undergo a full septic workup and be treated with empirical antibiotics.Measuring the appropriate markers for early sepsis might be beneficial, as mentioned in the setting of resuscitation guided by serum lactate[68,69]. However, this approach remains controversial in advanced liver disease because of: (1) The poor lactic acid metabolism in liver diseases; and (2) The weak association between hyperlactatemia and tissue hypoperfusion[70].
Managements of HRSIf renal function does not improve after adequate volume expansion, HRS-AKI and non-HRS-AKI must be in line with the differential diagnosis and be further assessed with simultaneous treatment. The goal of HRS-AKI treatment is to optimize the cardiac output and mean arterial blood pressure (MAP). Although there was no significant difference in outcomes between targeting the higher MAPs (80-85 mmHg)compared with the lower MAPs (65-75 mmHg) in septic AKI[71], the most recent study revealed that AKI incidence was lowest among those whose postresuscitation MAP was closest to or higher than their preadmission MAP[72]. Perhaps an individualized target MAP acts as a key marker for HRS-AKI. To increase the MAP and cardiac output, intravenous albumin administration along with systemic vasoconstrictors has shown promising outcomes. A number of systemic vasoconstrictors are used to counter splanchnic vasodilation, including a vasopressin analog (terlipressin)[73,74], an α-adrenergic agonist (norepinephrine)[75,76], and a combination of α-adrenergic agonist(midodrine) and somatostatin analog (octreotide)[77]. The most recent meta-analysis demonstrates that: (1) The treatment of HRS type 1 with terlipressin plus albumin attenuates the short-term mortality; and (2) Terlipressin plus albumin or noradrenaline plus albumin is superior to triple therapy with midodrine, octreotide and albumin[78]. Regarding AKI outcome, terlipressin infusion gives an earlier and stronger response than noradrenaline, with a higher reversal rate of HRS (40% vs 17%)and a lower rate of renal support requirement (57% vs 80%)[79]. Interestingly, theF

i
gure 2 Role of monocytes and macrophages in the immunological aspects of acute-on-chronic liver failure and acute kidney injury.Several risk factors addition to pre-existing chronic liver disease initiate hepatic inflammation which release various types of damage-associated molecular patterns, pathogen-associated molecular pattern, chemokines and inflammasomes. These mediators affect to the inflammatory cascade through monocyte and Kupffer cell activation which subsequently turn on either liver or systemic immunity. While, in the liver, Kupffer cells signal to bone marrow-derived monocytes for recruiting them to the liver,systemic (peripheral) monocytes also become activated monocytes which expanding the pro-inflammatory responses or systemic inflammatory response syndrome(SIRS). Overwhelming of pro-inflammatory cascade is supposed to be the background of acute kidney injury (AKI), similarly septic AKI (inflammation-related AKI).However, the functional reprogramming of both activated macrophages and activated monocytes could attenuate SIRS by differentiating to pro-restorative phenotypes that favors liver tissue resolution and healing. DAMP: Damage-associated molecular pattern; IL: Interleukin; PAMPs: Pathogens-associated molecular patterns; SIRS:Systemic inflammatory response syndrome; TNF-α: Tumor necrosis factor-alpha; AKI: Acute kidney injury.effectiveness of albumin depends on the cumulative administration dose, as a 100 g increase in cumulative albumin (max dose of 600 g albumin) increases the survival rate with a hazard ratio of 1.15 and a 95% confidence interval of 1.02-1.31[80]. Because most HRS-AKI develops in an ACLF setting and is associated with inflammatory mediators, therapeutic strategies not only for hemodynamic restoration but also for the attenuation of systemic inflammation might represent a paradigm shift in the treatment, as demonstrated by the balancing of monocyte function[81]. Recently,simvastatin, a well-known lipid-lowering agent, has demonstrated beneficial effects in an ACLF rat model with advanced chronic liver disease through the improvement of hepatic hemodynamics, microvascular dysfunction, and endotoxemia[82].
Roles of extracorporeal support systemsCompared with noncomplicated HRS-AKI, non-HRS-AKI and HRS-AKI in advanced ACLF have more morbidity and mortality because of their poor response to terlipressin and albumin. ACLF with ≥ 2 organ failures (ACLF grade 2-3) is associated with a 60%-75% rate of 28-d mortality[9], and therapies such as plasmapheresis that potentially ameliorate the ACLF severity by modulating the immune system may be an option. Currently, both renal and liver support in clinical studies have failed to have any survival advantage[83,84]. Regarding the mode of hemodialysis, continuous renal replacement therapy (CRRT) does not improve mortality in comparison with intermittent hemodialysis; however, CRRT might be well tolerated in patients with unstable conditions, including fulminant hepatic failure, as it does not raise intracranial pressure[85]. Indeed, the ADQI group recommends renal support only in cases with acute reversible components. Otherwise, renal support is not recommended in AKI-superimposed chronic liver disease, which is similar to the recommendation of limited utilization of liver support in liver transplantation[85].However, extracorporeal albumin dialysis may improve outcomes based on the clearance of excess bilirubin, bile acid, inflammatory cytokines, and endotoxins in systemic circulation[84]. The extracorporeal liver support is currently divided into 2 kinds of systems.
Cell-based liver support systems (bioartificial liver support systems):The data on bioartificial liver support systems benefit in patients with ACLF are still limited and from uncontrolled studies with limited numbers of patients.
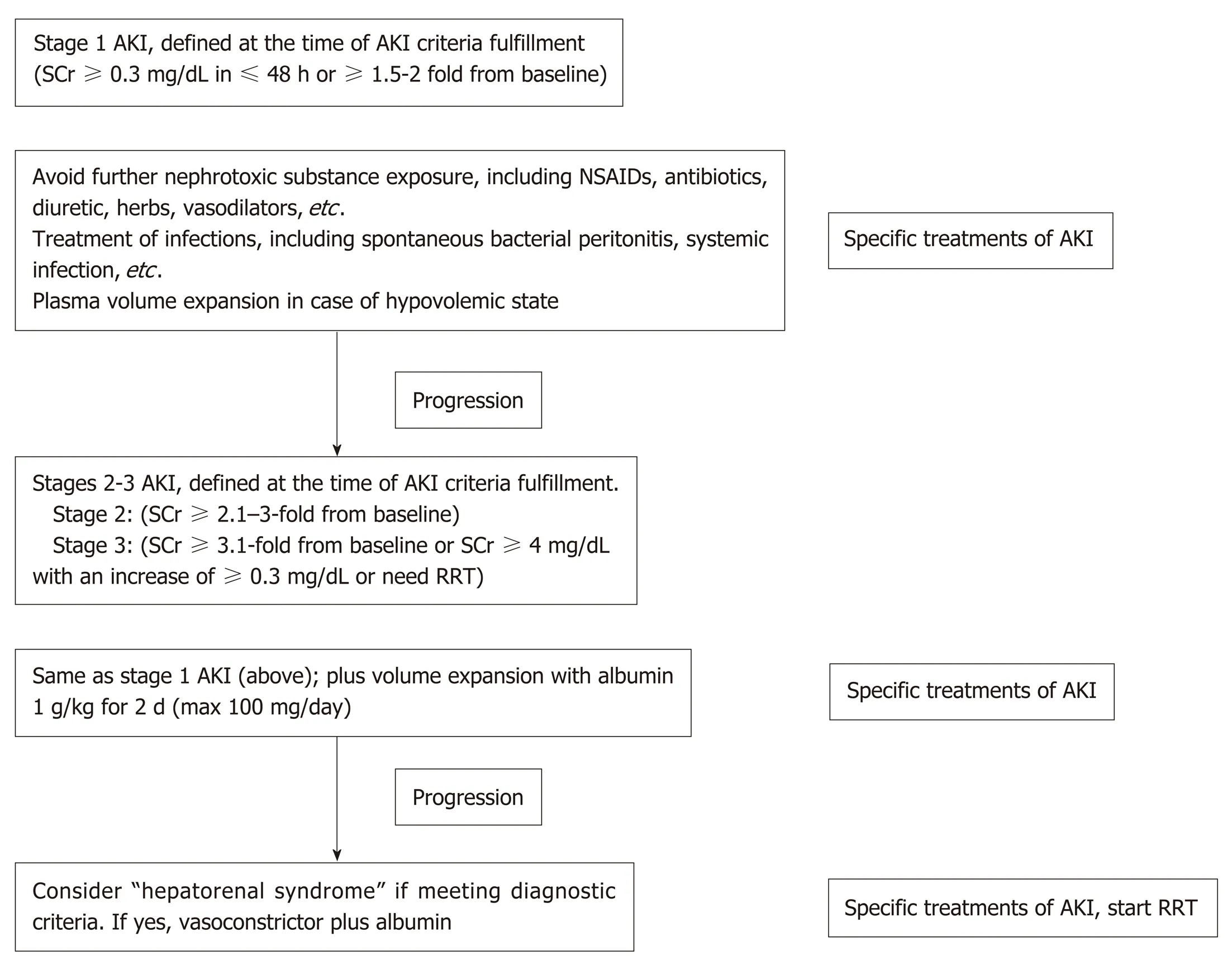
Figure 3 Summarized algorithm for the management of acute kidney injury according to the lnternational Club of Ascites-acute kidney injuryclassification, which combines Kidney Disease lmproving Global Outcomes criteria and conventional criteria in patients with cirrhosis and ascites.NSAIDs: Nonsteroidal anti-inflammatory drugs; SCr: Serum creatinine; RRT: Renal replacement therapy; AKI: Acute kidney injury.
Non-cell-based liver support systems:There are two major modalities regarding to the non-cell-based liver support systems-plasma therapy and albumin dialysis.Plasma therapy comprises 4 subtypes of techniques: (1) Standard plasma exchange is a simple circuit (Figure 4) that enhances the elimination of inflammatory cytokines and endotoxins by using 1.2 L of plasma as fluid replacement. However, it has so far failed to show a confirmed survival advantage; (2) High-volume plasma exchange(HVP). Unlike standard plasma exchange, the HVP procedure uses a large amount of fresh-frozen plasma (approximately > 10 L of fresh-frozen plasma or 15%-20% of ideal body weight) as replacement fluid. The established technique was first used for immunologically driven disorders in the early 1990s[86]. HVP decreases the vasopressor dose requirement in resuscitation and improves hepatic encephalopathy symptoms by decreasing blood ammonia and urea[87]. A recent prospective,randomized study was conducted in 183 patients with acute liver failure, with approximately 2.4 treatments per patient and 9 h per treatment, and it demonstrated a slightly improved survival by an intention-to-treat analysis (59% vs 48%) and reduced circulating proinflammatory mediators [IL-6, TNF-α, DAMPs[88]and soluble B7(CD80/CD89)[89]]. Unfortunately, there is no evidence on using HPV in ACLF so far.Further prospective studies are required to confirm these results in patients with acute liver failure and might be extended to patients with ACLF in the near future; (3)Plasma perfusion and bilirubin adsorption system and double plasma molecular absorption system, as shown in Figure 5. The fundamental mechanism in the plasma perfusion and bilirubin adsorption system is the separation of plasma that passes through an anion-exchange column (adsorbent), which has an adsorption effect on specific molecules (such as bilirubin, bile acid, and related similar molecular structures). Data from several studies showed a safe decrease in the plasma bilirubin concentration of approximately 18%-50% from baseline after one session[90,91]. The double plasma molecular absorption system is more sophisticated than plasma perfusion and bilirubin adsorption, combining both with an adsorbent for the reduction in inflammatory mediators, drugs or toxins (Figure 5). Although the rationale for using these systems seems to be that they will improve our understanding of ACLF pathophysiology, most studies on the double plasma molecular absorption system are in patients with hepatitis B-related liver failure[92-94].In our experience, the double plasma molecular absorption system decreased bilirubin and ammonia 25%-30% from baseline after one treatment session in patients with ACLF and cancer (unpublished data). The reduction in hyperbilirubinemia in patients with sepsis-related ACLF was also demonstrated in our case series(unpublished data); (4) Fractionated plasma separation and adsorption (FPSA)(Prometheus®). FPSA, first introduced in 1999, is fractionated through an albuminpermeable filter with a cutoff of 250 kDa. Albumin and other plasma proteins cross the membrane and pass across 2 columns in a series-an anion-exchange column and a neutral resin adsorber. The cleansed albumin/plasma is returned to the standard blood pool circuit, where it is then treated by conventional high-flux hemodialysis(Figure 6). Clinical studies have evaluated the effect of FPSA in ACLF patients, with favorable outcomes, indicating that the use of FPSA is well tolerated and decreases the circulating levels of serum bilirubin, bile acids, and ammonia[95-100]. However, the improvement of neurological status and hemodynamics following FPSA treatment is controversial[96,98]. A prospective, randomized clinical trial (HELIOS study)[100]conducted in 145 ACLF patients demonstrated that FPSA decreased serum bilirubin with nondifferent 28-d survival rates (66% vs 63%). The secondary endpoint showed a significant improvement in survival in ACLF patients using the cutoff of Model for End-Stage Liver Disease (MELD) score > 30. It should be emphasized that there is no further confirmatory study or any study in an acute liver failure setting so far.
Albumin dialysis has been classified as molecular adsorbent recycling system(MARS) and single plasma albumin dialysis (SPAD). Technically, MARS consists of 3 main circuits of the blood delivery system, albumin flow, and routine hemodialysis system (Figure 7). In the MARS circuit, blood is dialyzed through an albuminsaturated high-flux dialyzer with 20% human albumin (600 mL) and is used as the dialysate for carrying the albumin-bound toxins into the dialysate, where the watersoluble and albumin-bound toxins will be removed from albumin through 2 sequential adsorbent columns containing activated charcoal and anion-exchange resin. Then, the toxin-free albumin will be circulated in the circuit. The highmolecular-weight toxins (> 50 kDa) are not removed by MARS due to the dialyzer pore size limitation. The RELIEF study is the largest randomized trial using MARS in ACLF, with 189 patients from 19 European centers, and it demonstrated that undergoing up to ten 6-h to 8-h sessions of MARS was associated with the improvement of hepatic encephalopathy (from grade 2-4 to grade 0-1; 63% vs 38%,respectively; Ρ = 0.07) without a 28-d survival benefit[101]. Similarly, the FULMAR trial was a randomized controlled study using MARS in 102 patients with acute liver failure fulfilling transplant criteria and demonstrated no significant differences in 6-month survival between the MARS (85%) and standard medical treatment (76%)groups (Ρ = 0.28)[102]. However, the major confounder in the study was the short transplant waiting time [median 16.2 h, mostly within 24 h (75%)], which might be too short to see the benefit of liver support. Interestingly, neither MARS nor FPSA showed significant decreases in inflammatory markers (IL-6, IL-8, IL-10, and TNF-α)after the treatment session in a small study[103].
In contrast to MARS, SPAD uses a standard CRRT system without any additional columns (Figure 8). After a SPAD cycle is run, water-soluble toxins (ammonia,creatinine, urea, uric acid) are removed almost completely. Blood is dialyzed against a standard dialysis solution with the addition of albumin to the dialysate. The most effective albumin concentration was 3%. A further increase in the albumin concentration to 4% did not lead to a significant increase in the detoxification[104].However, SPAD increased the detoxification efficiency of albumin-bound substances from 350 mL/h to 700 mL/h (for bilirubin) or 1000 mL/h (for bile acids) of the dialysate flow in SPAD for the first time[104]. Unfortunately, SPAD has been evaluated in a case-control fashion mostly in drug overdose-related liver failure, with controversial outcomes[105-107].
Roles of liver transplantationSo far, liver transplantation is the only treatment modality for the reversal of either HRS-AKI or non-HRS-AKI in an ACLF setting. Recently, a large retrospective study of liver transplantation in patients with either HRS-AKI or non-HRS-AKI in ACLF demonstrated that 1-year and 5-year survival rates were 91% and 77%, respectively.However, those with a renal injury background of either diabetes or hypertension showed a lower survival rate (1-year and 5-year survival at 87% and 71%,respectively)[108]. Pretransplant AKI severity contributed as an important factor for renal function recovery at 4-6 wk posttransplantation. Thus, simultaneous liverkidney transplantation should be considered in those with predicted renal recovery longer than 6 wk[85].

Figure 4 Plasma exchange circuit.
Roles of the novel monocytes and macrophage modulatorsRegarding the high rate of mortality and limited treatment options for ACLF and AKI following ACLF; novel, robust therapies are needed.
Targeting liver macrophage:So far, unfortunately, none of proven macrophagedirected therapies has been recommended in ACLF although a number of immunomodulators including N-acetylcysteine, albumin and glucocorticoids are approved for other immune-related liver diseases[46]. The limitation of knowledge is probably due to the limitation in retrieving human liver tissue and the differentiation process for liver macrophage in human[46].
Targeting inhibition of Kupffer cell activation:Inhibition of progression of the early phase of SIRS in ACLF could be attenuate the signal induction for Kupffer cell activation especially targeting to inhibit released DAMPs. Recent study demonstrated potential beneficial of HMGB-1 blockade treatment in rat with ACLF model through alleviating inflammation in SIRS[109]. Likewise, the blocking responses against of histones (a DAMP) attenuated cytokine production and reduced liver injury severity[110]. In addition, the prevention and treatment of bacterial translocation by appropriate antibiotics is the most effective attenuation of initial innate immune activation leading to the subsequently Kupffer cell inhibition[111].
Targeting inhibition of monocyte recruitment into the liver:The recruitment of monocytes into liver is mediated mainly through chemokine system including; CCL2-CCR2 and CCL5-CCR5 for the recruitment of monocyte and lymphocyte,respectively[112]. As such, Cenicriviroc, an CCR2/CCR5 inhibitor, in a randomized,double-blind study in 289 patients with NAFLD and hepatic fibrosis demonstrated a reduction on fibrosis in treatment group (n = 145) compares with placebo (n = 144)(20% vs 10%) without the reduction of the primary endpoint (NAFLD activity score)[113]supporting the animal model data[112].
Targeting promotion of macrophage differentiation:An early promotion of macrophage differentiation from activated macrophages phenotype into a prorestorative phenotype (such as by corticosteroids) might enhance the resolution of liver injury[111]. However, the data on macrophage differentiation treatment in ACLF is still too less. Corticosteroids might provides some advantages in the early phase of ACLF but possibly augment infectious complications in the late phase[114].
Roles of the novel granulocyte colony-stimulating factor therapyACLF is mainly mediated through immune dysfunction and is also susceptible to the serious infections. Accordingly, treatment by hematopoietic growth factor to induce immune cells seems reasonable to restore immune homeostasis through improving impaired phagocytosis[115]and mobilization of CD34+ progenitor cells into liver[116].The first randomized placebo-controlled study in 2012 using granulocyte colonystimulating factor (G-CSF) in patients with ACLF demonstrated an improved 60-d survival rate and a reduction in multiorgan injury on the basis of clinical scores,including those of the MELD and Sequential Organ Failure Assessment (66% vs 26%)[117]. Likewise, G-CSF improved the 90-d mortality rate (78% vs 30%) in a randomized trial on 46 patients with ACLF[118]. The largest study, on 55 patients with hepatitis B-related ACLF randomized to receive standard medical treatment or G-CSF in addition to standard medical treatment, again showed a significant benefit in the 90-d survival rate in the G-CSF-treated group (48% vs 28%)[119]. Interestingly, the most recent randomized study in 32 patients with hepatitis B virus-ACLF showed that 5 μg/kg/day of G-CSF for six consecutive days in addition to the standard treatment,improves survival, facilitates clinical recovery, prevents renal failure and protects from hyponatremia[120].

Figure 5 Plasma perfusion and bilirubin adsorption system and double plasma molecular absorption system circuit. DPMAS: Double plasma molecular absorption system.
CONCLUSION
AKI following advanced liver cirrhosis is a critical condition. The early recognition and rapid diagnosis of AKI in these patients may improve therapeutic outcomes.However, the understanding of the pathogenesis and the quality of the diagnostic tools for the simultaneous injury of the kidney and liver are still limited. A distinct diagnostic criterion for differentiating the points on the prerenal-HRS-ATN spectrum needs robust validation as well as an accurate distinction between HRS-AKI and non-HRS-AKI in ACLF. Inflammation is increasingly recognized as an important driver of AKI, particularly in patients with infection and multiorgan failure. As AKI in chronic liver disease is persistent and rapidly becomes irreversible by medical treatment as the condition prolongs, novel therapies and new approaches for either liver or renal support are required.

Figure 6 Fractionated plasma separation and adsorption (Prometheus®) circuit.

Figure 7 Molecular adsorbent recycling system circuit. MARS: Molecular adsorbent recycling system.

Figure 8 Single-pass albumin dialysis circuit.
 World Journal of Gastroenterology2019年28期
World Journal of Gastroenterology2019年28期
- World Journal of Gastroenterology的其它文章
- Systematic review of nutrition screening and assessment in inflammatory bowel disease
- Comparison of the use of wireless capsule endoscopy with magnetic resonance enterography in children with inflammatory boweldisease
- Effect of low-dose aspirin administration on long-term survival of cirrhotic patients after splenectomy: A retrospective single-center study
- Comparison of outcomes between complete and incomplete congenital duodenal obstruction
- MiR-205 mediated APC regulation contributes to pancreatic cancer cell proliferation
- Novel technique for endoscopic en bloc resection (EMR+) -Evaluation in a porcine model