
表观遗传药物研发的现状与挑战
2019-08-08 03:32:10江芮吕柯孬潘学峰崔新霞申世刚丁良
生物技术通报 2019年8期
江芮 吕柯孬 潘学峰,, 崔新霞 申世刚 丁良
(1. 河北大学医学院,保定 071000;2. 北京理工大学生命学院,北京 100081;3. 河北大学化学与环境学院,保定 071000)
表观遗传机制主要涉及染色质中DNA、组蛋白等分子的化学修饰状态对基因表达的影响,及基因所编码的产物(生物分子)所决定的遗传性和不可遗传性改变。当前,已知的表观遗传修饰主要包括DNA和组蛋白的修饰,如DNA甲基化/去甲基化、各种类型的组蛋白可逆性修饰,以及调节非编码RNA等。上述表观遗传修饰可通过影响染色质的构象状态影响基因转录,如出现在人和其他哺乳类生物基因组中“CpG岛”内胞嘧啶甲基化可使相应部位染色质变 “致密”,抑制基因转录;组蛋白N端尾部无序结构域内氨基酸残基的乙酰化修饰可削弱组蛋白和DNA之间的作用,促进基因转录。迄今为止,在包括癌症、心脏病、糖尿病和精神疾病等长期困扰人类健康的重大疾病过程均发现有异常的表观遗传修饰[1]。利用“药物”人为干预这些疾病过程中的异常基因表达(基因沉默或上调)以实现对疾病的治疗已成为当今医药科技的一个热点。基于此,本文试图对当前表观遗传药物研发的现状及存在的问题进行分析和总结。
1 表观遗传修饰类型
1.1 DNA甲基化修饰
哺乳动物DNA甲基化修饰通常由DNA甲基转 移 酶(DNA methyltransferase,DNMT) 将 S-腺苷甲硫氨酸(SAM)的“甲基”添加到位于基因上游启动子或结构基因的5'侧的CpG岛胞嘧啶中[2](图1-A)。目前已发现4种DNMTs,其中DNMT1负责基因组特定部位的DNA甲基化模式的维持,DNMT3A、DNMT3B以及DNMT3L则负责胚胎早期发育过程所必需的DNA甲基化修饰[3]。CpG岛甲基化修饰通常会关闭所在部位基因的转录。研究表明,一些肿瘤细胞内抑癌基因的失活可归因于其启动子区CpG岛的过度甲基化修饰和基因组范围内低水平甲基化修饰。这种DNA的甲基化修饰紊乱可以直接诱发细胞转化,增加罹患肿瘤风险[4]。与此类似,诸多神经退行性疾病也存在异常DNMT活性(突变导致)[5]。当今,DNA甲基化的差异性已经被用作肿瘤发展的早期诊断依据,如CpG岛甲基化表型(CpG Island methylator phenotype,CIMP)已用于不同亚型的胃癌的表征[6]。
1.2 组蛋白修饰
核小体核心组蛋白N端无序结构域中赖氨酸、精氨酸残基部位的乙酰化/去乙酰化、甲基化/去甲基化、泛素化修饰,以及丝氨酸残基部位的磷酸化修饰等是组蛋白表观修饰的主要形式[7](图1-B)。其中,甲基化和乙酰化是最重要的组蛋白修饰方式。组蛋白修饰可影响核小体的稳定,与异染色质形成、DNA损伤修复、基因组稳定性等直接有关[8]。组蛋白修饰异常可见于肿瘤、心血管疾病、神经变性疾病和糖尿病等常见疾病[9]。
1.3 非编码RNA
非编码RNA(Non-coding RNA,ncRNA)是不编码蛋白质的RNA分子的统称,即除mRNA之外所有类型的RNA分子。可依据所含碱基数分为小非 编 码 RNA(Small noncoding RNAs,microRNA,miRNA)和长非编码RNA(Long noncoding RNAs,lncRNA)[10]。其中miRNA参与基因表达的调控(截至2018年10月,剑桥MicroRNA数据库(miRBase)已有38 589个MicroRNA条目收录(The miRBase Sequence Database,Release 22.1),而 lncRNA 则参与异染色质形成、X染色体失活、基因组印记、细胞凋亡等过程[11]。目前,临床和实验室研究最多的是小干扰 RNA(Small interfering RNA,siRNA)。SiRNA通过与靶基因的mRNA(Messenger RNA)特异结合可有效降解特定的mRNA,造成相应基因沉默(RNA 干扰)[12](图 1-C)。2018 年 8 月,美国食品和药物管理局(FDA,USA)批准了第一种基于RNA干扰的治疗方法,用于治疗一种可能损害心脏和神经功能的罕见疾病[13]。
2 表观遗传药物的研究
2.1 DNA甲基转移酶抑制剂
针对异常DNA甲基化研发的DNA 甲基转移酶抑制剂(DNMTsI)主要分为核苷类抑制剂、非核苷类抑制剂和反义寡核苷酸类抑制剂。
2.1.1 核苷类抑制剂 核苷类DNMTsI通过在DNA复制过程中掺入新合成的DNA分子发挥作用。比如用相应的DNMTsI取代“CpG”岛中的胞嘧啶,在不影响互补链间“CG”配对的同时,避免DNMT对原有胞嘧啶碱基的识别和甲基化修饰。这类抑制剂包括阿扎胞苷(5'-氮杂胞苷,Azacitidine)、地西他滨(Decitabine)、泽布拉林(Zebularine)等或以胞嘧啶核苷为母体的衍生物等。这些核苷类DNMTsI能与DNMT中半胱氨酸残基上的巯基共价结合的方式抑制 DNMT 的转甲基活性[14](表 1)。
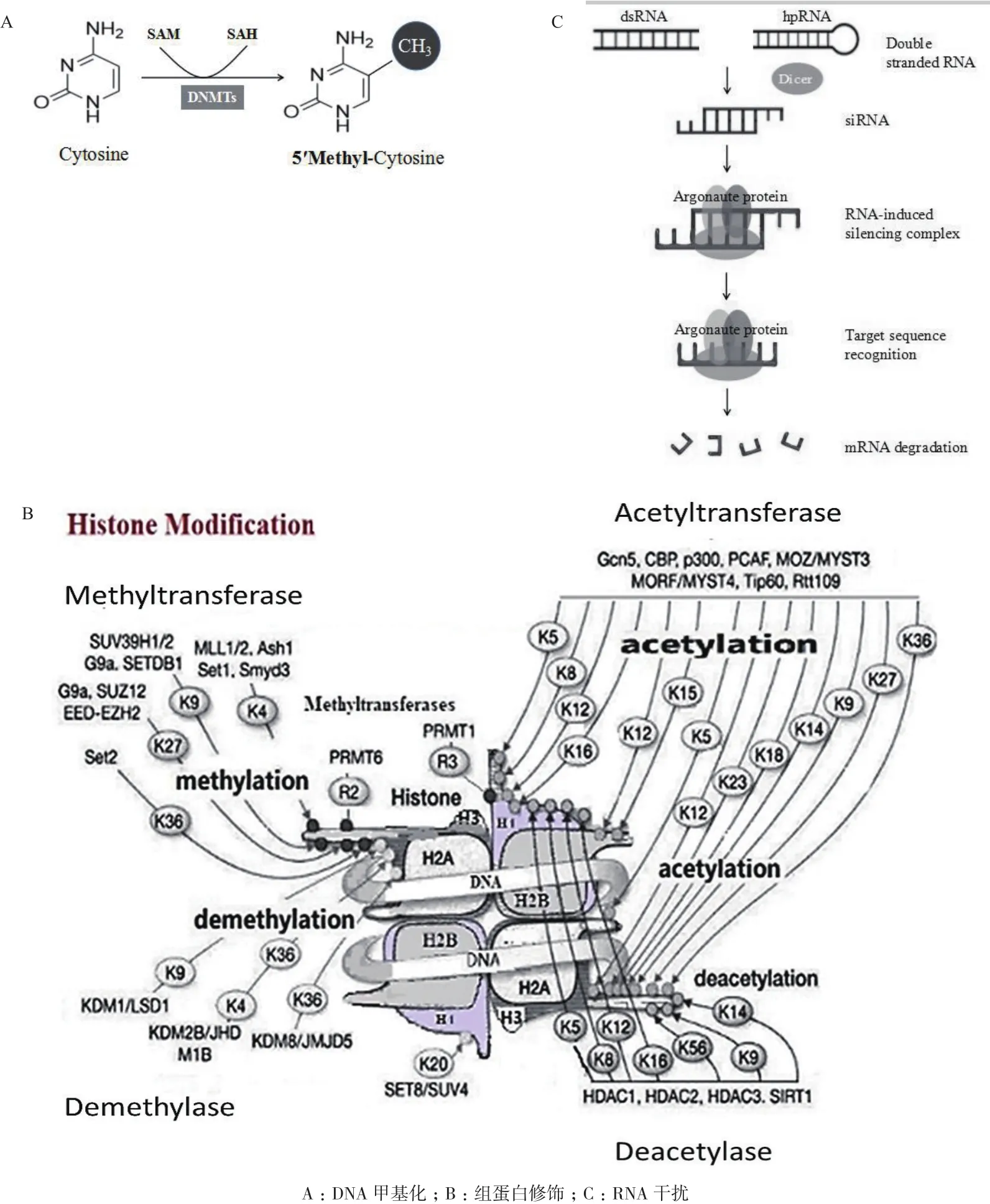
图1 表观遗传修饰的类型方式
2005年,美国FDA批准了阿扎胞苷和地西他滨在骨髓增生异常综合征(Myelodysplastic syndromes,MDS)等恶性血液系统疾病的临床治疗[15-16]。但上述药物的临床治疗剂量常导致患者出现严重胃肠道反应和表现出骨髓毒性[17-19]。当前研究者正对上述药物的化学结构进行修饰,以期获得稳定性好、对肿瘤细胞的选择性高、药物不良反应更低、且水溶性好、便于口服给药的新型衍生物。此外,临床上常见的硫鸟嘌呤(6-Thioguanine)可通过与DNA转甲基酶形成共价复合物抑制 DNMT1的活性(也抑制胞苷脱氨酶)[20],常用于治疗急性淋巴细胞白血病,自身免疫性疾病(如克罗恩氏病、类风湿关节炎)和器官移植接受患者;泽布拉林(Zebularine)则是一种含有2-(1H)嘧啶环酮的胞苷类DNA甲基化抑制剂,能通过与DNMT共价结合有效解除处于超甲基化修饰状态的p16基因的DNA甲基化修饰[21]。概言之,核苷类DNMT抑制剂一般均能与DNMT酶共价结合,以复合物形式影响DNA的甲基化修饰,因此这类药物普遍存在细胞毒性。临床中,为了降低药物的副作用一般采取与其他类抗肿瘤药物联合用药。
2.1.2 非核苷类DNA甲基转移酶抑制剂 非核苷类DNMTsI不含胞嘧啶核苷的骨架结构,依据化学结构分为氨基苯甲酸类、多酚类、肼类、邻苯类、二酰胺类等[22-24]。其中,氨基苯甲酸类的代表是普鲁卡因(Procaine),其能特异性结合富含“CpG”的DNA序列,阻止DNMT与该DNA部位结合。普鲁卡因可在多种肿瘤细胞中发挥阻止DNA甲基化作用[25]。多酚类药物包括表没食子儿茶素没食子酸酯[(-)-epigallocatechin-3-gallate,EGCG,绿茶的一种多酚类化合物]能与DNMT1的催化中心结合,干扰DNMT对 “CpG”中胞嘧啶甲基化修饰[25-26]。姜黄素(Curcumin)也是一种多酚类化合物,可选择性抑制DNMT1、组蛋白乙酰基转移酶p300(Histone acetylatransferase,IC50为 50-25 μmol/L)和组蛋白去乙酰基酶(Histone deacetylase),且能激活Nrf2-Keap1途径(清除氧游离基),抑制NF-κB(Nuclear factor-kappa B,NF-κB)的激活(降低炎性细胞因子的表达)[27]。有研究表明,姜黄素可借抑制NF-κB以时间和剂量依赖的方式抑制胰腺癌细胞的生长[28]。尽管姜黄素安全性好,但水溶性差,体内代谢快,因此其生物利用度低[29]。根据2017年进行的针对120多项有关姜黄素研究的回顾分析可知,姜黄素的不稳定、高活性及难于被有效利用等是制约姜黄素临床应用的主要问题。当前美国政府已经为“替补和整合健康国家中心”(The National Center for Complementary and Integrative Health)拨款1.5亿美元用于姜黄素的研究[30]。其中,通过改进姜黄素递送方式如制成纳米颗粒或脂质体以提高其生物利用度成为了研究重点[31]。很多案例表明,人工设计的抑制剂似乎优于天然来源的抑制剂,如RG108可直接抑制DNMT催化结构域,且检测不出任何细胞毒性[32]。RG108是依据DNMT催化结构域的分子结构,利用计算机模拟优化得到的第一个人类DNMT抑制剂。最近,随着越来越多的DNA甲基转移酶结构域晶体结构解析成功,将会有更多人采取类似RG108的方法来设计和优化DNMT抑制剂[33]。同时,基于线性判别分析(Linear discriminant analysis,LDA)及虚拟筛选可能会助推针对天然来源的DNMT抑制剂的有效识别。最近,已利用高通量筛选(High throughput screening,HTS)发现了2种新的 DNMT 抑制剂 SW155246 和 SID49645275[34],但这些筛选出的DNMT抑制剂的药理学信息未见公开。
2.1.3 反义寡核苷酸类抑制剂 反义寡核苷酸(Antisense Oligonucleotide)可与mRNAs某一区段互补发挥抑制基因表达的作用,如人工合成的反义寡核苷酸药物MG98可特异结合DNMT1 mRNA3'端,抑制其作为蛋白质翻译的模板,可有效激活因甲基化而沉默的抑癌基因,从而发挥抑制肿瘤的作用[35]。
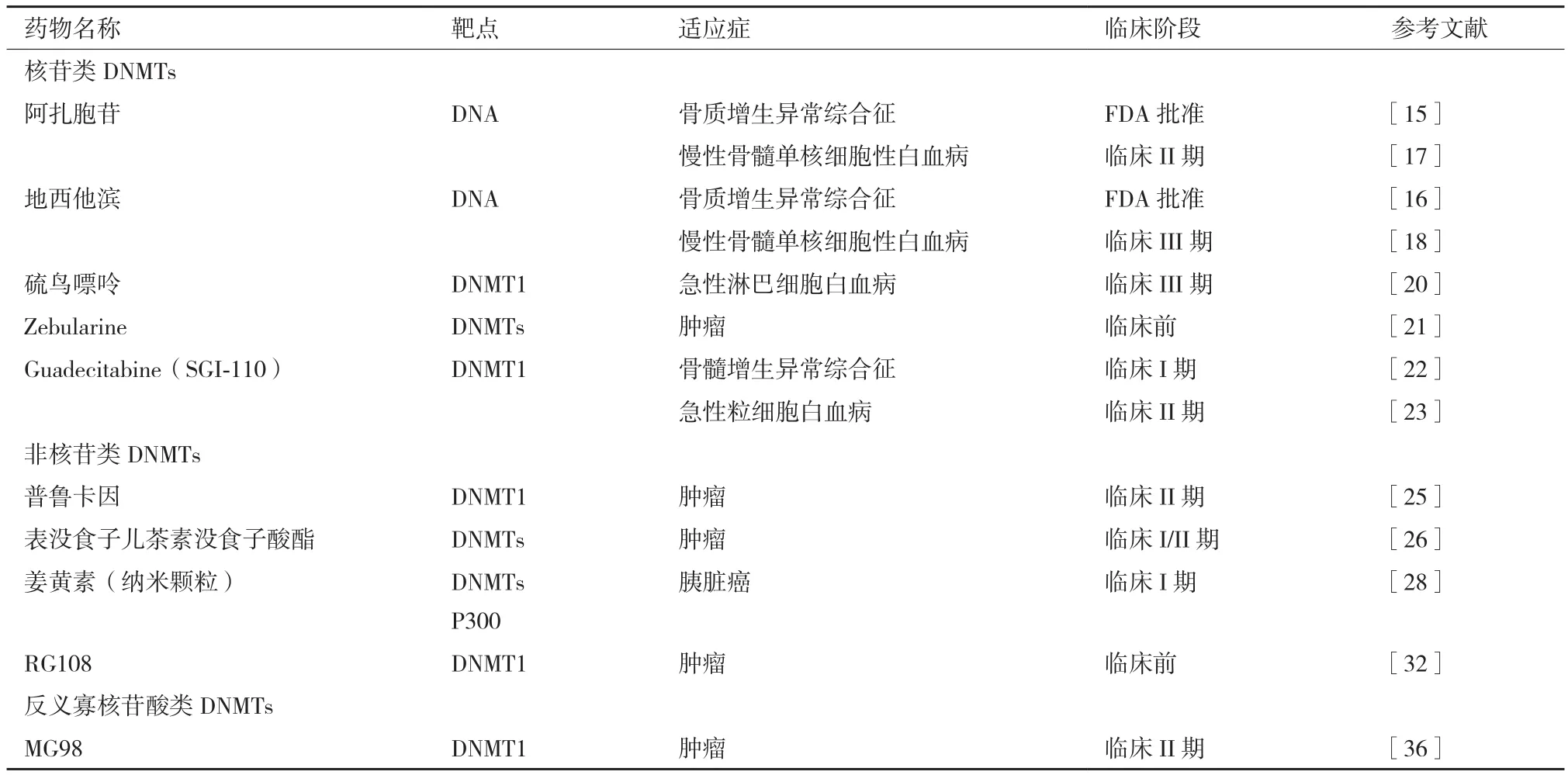
表1 DNA甲基转移酶抑制剂(DNMTsi)的临床研究阶段
2.2 组蛋白靶点抑制剂
当前,处于临床试验中的组蛋白靶点抑制剂主要为组蛋白甲基转移酶抑制剂和组蛋白去乙酰化酶抑制剂,其他类型的组蛋白靶点抑制剂相对较少(表 2)。
2.2.1 组蛋白甲基转移酶抑制剂 组蛋白甲基化修饰主要发生在组蛋白赖氨酸和精氨酸残基上,分别由组蛋白赖氨酸甲基转移酶(Histone lysine methyltransferases,HKMTs)与组蛋白精氨酸甲基转移酶(Histone arginine methyltransferase,HRMTs)负责催化。HKMTs催化赖氨酸残基的单、双和三价甲基化,而HRMTs催化精氨酸的单甲基化、不对称或对称二价甲基化[37]。
迄今,已鉴定和表征了50多种HKMTs[38]。其中,PCR2(Polycomb repressive complex 2)通过对组蛋白H3赖氨酸K27 位点甲基化修饰沉默基因转录[39]。针对PCR2的催化结构域EZH2(Enhancer of zeste homolog 2)研发了系列组蛋白甲基转移酶抑制剂(HMTsi),如 Tazemetostat(EPZ-6483)可有效抑制EZH2的催化活性或阻碍EZH2的SET区域发生改变,对抑制非霍奇金淋巴瘤(Non-Hodgkin lymphoma,NHL) 有 效[40], 但 2018年 9月,FDA 叫 停 了Epizyme公司正在进行的关于Tazemetostat临床实验,原因是需要对由Tazemetotstat可能诱发包括T细胞淋巴母细胞淋巴瘤(T-LBL)等继发性恶性肿瘤的风险加以综合评估(表2)。GSK126是新型的EZH2抑制剂,可高效抑制H3K27me3及H3K27me2,同时能有效抑制EZH2突变型DLBCL细胞系的增殖,并诱导敏感细胞系中EZH2靶基因的转录激活[41]。此外,GSK126还可通过阻断Wnt/β-catenin信号通路,消除干细胞样骨髓瘤细胞[41]。据报道,骨髓瘤细胞的增殖与H3K27甲基化有关[42]。同时,组蛋白甲基转移酶MMSET过表达能促进多发性骨髓瘤的发生,与患者的预后、肿瘤的分期分级和侵袭性密切相关[43]。
G9a( 又称 EHMT2 或 KMT1C) 和 GLP(G9a样蛋白1,也称为EHMT1或KMT1D)是主要的赖氨酸甲基转移酶,主要催化H3K9双价甲基化修饰,以及H3K27 的甲基化修饰等[44]。研究发现G9a在食管鳞状细胞癌、肝细胞癌、侵袭性肺癌、脑癌、多发性骨髓瘤和侵袭性卵巢癌中过表达[45]。BIX01294是第一个能选择性抑制G9a/GLP的底物竞争性HKMTs抑制剂,能下调H3K9me2表达。然而BIX01294在细胞测定中活性较弱,且在浓度高于 4.1 μmol/L 时具有细胞毒性[37]。UNC0638是基于UNC0224和UNC0321进一步合成出的化合物,在多种细胞内能特异地大幅度下调H3K9me2。但UNC0638在体内药代动力学较差,不适合动物研究[46]。进一步优化合成出UNC0642,其不仅表现出较高的体外效能和优异的选择性,且在细胞中表现出强大的靶向性[46]。端粒沉默干扰因子(Disruptor of telomeric silencing1-like,DOT1L)主要催化H3K79的单、双和三价甲基化修饰(H3K79me1,H3K79me2 和H3K79me3),是体内融合基因MLLAF9诱导白血病发生和维持所必需的HMTs[47]。有报道表明,当前已发现的DOT1L抑制剂均存在代谢不稳定问题[48]。
虽然HKMTs抑制剂在临床前和临床试验中表现出抗肿瘤作用,但当前大多数的HKMT抑制剂都缺乏细胞和动物水平上药理学信息。HRMTs迄今在哺乳动物中共发现了9种[50],HRMTs抑制剂多停留在细胞生物学水平上,尚未见体内实验研究报道。
2.2.2 组蛋白去甲基化酶抑制剂 组蛋白甲基化的动态修饰在染色质重塑过程中发挥至关重要的作用,错误的组蛋白去甲基化与众多人类疾病有关[51]。自2004年首次发现组蛋白去甲基化酶(Histone demethylase,HDMs)以来,已确定了2大类组蛋白去甲基酶,分别为赖氨酸特异性组蛋白去甲基酶(Lysine specific demethylases,LSDs)和 JMJD(Jumanji domain-containing protein)组蛋白去甲基化酶[52]。由于组蛋白的甲基化是一种动态可逆的表观遗传修饰过程,因此人们相信组蛋白中大多数能被甲基化修饰的赖氨酸残基都存在着专用的组蛋白去甲基酶。当前,针对这些组蛋白赖氨酸去甲基酶抑制剂的研究已经取得了不少的成果。例如,GSK-J1是一种高效的 H3K27去甲基酶抑制剂,体外试验显示针对含有JMJD3(KDM6B)和 UTX(KDM6A)结构域的组蛋白去甲基酶的 IC50分别为 28 nmol/L 和 53 nmol/L,而且对这两类组蛋白去甲基的选择性高出针对其他类型的组蛋白去甲基10倍以上[46]。GSK-J4盐酸盐是GSK-J1前体,具有细胞渗透能力,而且是第一个可选择性抑制H3K27组蛋白去甲基酶JMJD3和UTX的抑制剂。已有的数据表明,GSK-J4盐酸盐体外无细胞试验时的IC50为60 nmol/L,能选择性抑制JMJ家族的去甲基化酶活性[53]。AS-8351是一种可被用于诱导人胚肺成纤维细胞向功能性心肌细胞的重编程过程的HDMs抑制剂[54]。ORY-1001(RG-6016)二盐酸盐是对赖氨酸特异性组蛋白去甲基酶LSD1/KDM1A具有较强选择性的抑制剂,IC50小于20 nmol/L,同时对FAD依赖的氨基氧化酶也具高度选择性,因此较为安全,可口服给药[55]。SP2509则是最近研发的一种对组蛋白赖氨酸残基去甲基化酶LSD1具有选择性的抑制剂[52]。SP2509能在体外实验中降低共阻遏物CoREST与LSD1的结合,促进启动子区 “转录许可型”(Transcription permissive)H3k4me3修饰,提高慢粒白血病患者细胞内p21、p27及与“CAAT/增强子”结合的蛋白α的表达,促进慢粒白血病细胞的分化和抑制其克隆性生长。经过尾静脉注射可有效延长免疫剥夺小鼠的存活[56]。同时,SP2509能有效抑制人急性髓细胞性白血病(Acute myeloid leukemia,AML)细胞的增殖,并诱导其凋亡[57]。LDS1在各种血液系统的恶性肿瘤中呈现高表达,其通过促使抑癌基因p53位点上的K370me去甲基化,从而抑制p53的信号传递[58]。
尽管如此,现有在研的HDMs抑制剂普遍存在活性低等问题。造成这种情况的根本原因可能与细胞内HDMs所能影响的表型过于复杂,造成HDMs的确切作用机制难以精确辨认,以及尚缺乏更详尽的HDMs非催化结构域功能信息有关。
2.2.3 组蛋白去乙酰化酶抑制剂 组蛋白去乙酰化酶(Histone deacetylase,HDACs)是催化组蛋白去乙酰化的一类蛋白酶,在基因表达的表观遗传调控中发挥不可或缺的作用。迄今已在哺乳动物中发现了至少18个HDACs的亚型,可依据酵母菌HDAC序列的相似性分为4类[59]。针对上述组蛋白去乙酰化酶的抑制剂(HDACIs)具有诱导肿瘤细胞损伤、细胞周期停滞,增强bax基因表达抑制bcl-2的基因表达,以及影响细胞周期抑制因子p21CIP1/WAF1等作用,在体外细胞培养和动物模型中均被证明具有抗肿瘤作用[60]。目前研究最广泛的HDACIs主要分为异羟肟酸类、苯甲酰胺类、环肽类、短链脂肪酸类和多酚类。当前市面上有很多HDAC抑制剂销售,但主要用于实验研究。其中临床效果较好的有曲古抑菌素(Trichostatin,TSA)、CI994等。曲古抑菌素是最早发现的天然可逆性HDACIs,通过抑制Ⅰ型和Ⅱ型HDACs可有效上调细胞中组蛋白乙酰化水平,促进细胞分化,阻滞细胞周期,诱导肿瘤细胞凋亡[61]。以喉鳞癌为例,HDAC1与SIRT1(HDACs之一)在喉鳞癌组织中呈现高表达,其与淋巴结转移和预后相关[62]。喉鳞癌属于鳞状细胞癌,其通过HDAC1和HDAC2与p53相关蛋白p63(TP63)结合形成活性转录抑制因子复合物,抑制促凋亡Bcl-2基因,促进鳞状细胞癌的发展[63]。CI994(Tacedinaline)目前主要用做抗癌药,对HDAC1的抑制IC50为0.57 μmol/L,可使细胞周期停滞在G1 期[88]。
当前,尽管发现了众多HDAC抑制剂,但普遍选择性不高。诸多研究表明HDAC各亚型在生长发育及疾病的发生发展过程发挥重要作用[8],因此当前亟需高效的HDAC抑制剂,但由于各种亚型催化位点序列的高相似性,使得高选择性HDAC抑制剂的设计、合成变得异常困难。
2.2.4 组蛋白乙酰化转移酶抑制剂 组蛋白乙酰转移酶(Histone acetyltransferases,HAT)在组蛋白、转录因子、核受体和酶等细胞蛋白的赖氨酸残基上“加载”乙酰基,激活基因转录。这类乙酰基转移酶不只局限于对组蛋白乙酰化修饰,也能对许多非组蛋白进行乙酰化修饰。HAT为双底物酶,能催化辅因子乙酰辅酶A(Ac-CoA)和含赖氨酸的两个底物之间的反应,根据序列同源性和底物乙酰化性质,现已鉴定出HAT1、GCN5/PCAF、MYST、p300/CBP和Rtt109等5个亚家族[89]。HAT抑制剂(HATsi)主要为双底物抑制剂和小分子抑制剂。双底物抑制剂能模拟两种HAT底物,但其代谢稳定性差,细胞渗透性不足,限制了在细胞中的应用;当前已有的小分子HAT抑制剂主要来自天然产物,其抑制剂不具选择性,且大部分含有酚类结构,易于氧化[90]。如来自银杏果实和叶子中的有毒酚类化合物银杏酸原先发现可有效抑制蛋白的“sumoylation”化,现发现其也能抑制组蛋白乙酰化转移酶(HAT)[91]。与此类似,茴香酸(Anacardic acid)也能有效地抑制p300及p300/CBP相关因子的组蛋白乙酰基转移酶活性,但除此之外,茴香酸还能抗细菌和抗微生物,抑制前列腺素合成,以及抑制酪氨酸酶和脂肪氧合酶等活性[92]。除天然的组蛋白乙酰基转移酶抑制剂之外,人工设计的组蛋白乙酰基转移酶抑制剂也陆续上市,如A-485、C646和Remodelin等。A-485是可选择性抑制组蛋白乙酰基转移酶p300/CBP的组蛋白乙酰基转移酶抑制剂,对p300 HAT的IC50为0.06 μmol/L[93]。相较于BET溴区蛋白和其他多于150种的非表观遗传药物靶点,A-485对p300/CBP更具选择性。由于p300/CBP参与结肠癌、肝癌、前列腺癌等在内的多种人类恶性肿瘤的发生发展,尤其是血液系统恶性肿瘤,因正常造血干细胞的产生和功能发育过程中需要p300/CBP,其失活突变会造成血液系统恶性肿瘤的发生[94]。A-485在大多数多发性骨髓瘤(MM)细胞系中能有效抑制其生长[95]。与此相似,C646也是一种组蛋白乙酰基转移酶p300的抑制剂,相比于其他乙酰基转移酶,C646可优先作用于p300,能阻断胃癌细胞系的存活和侵袭[96]。此外,“Remodelin”是一种有效的针对乙酰基转移酶 NAT10 的抑制剂[97]。
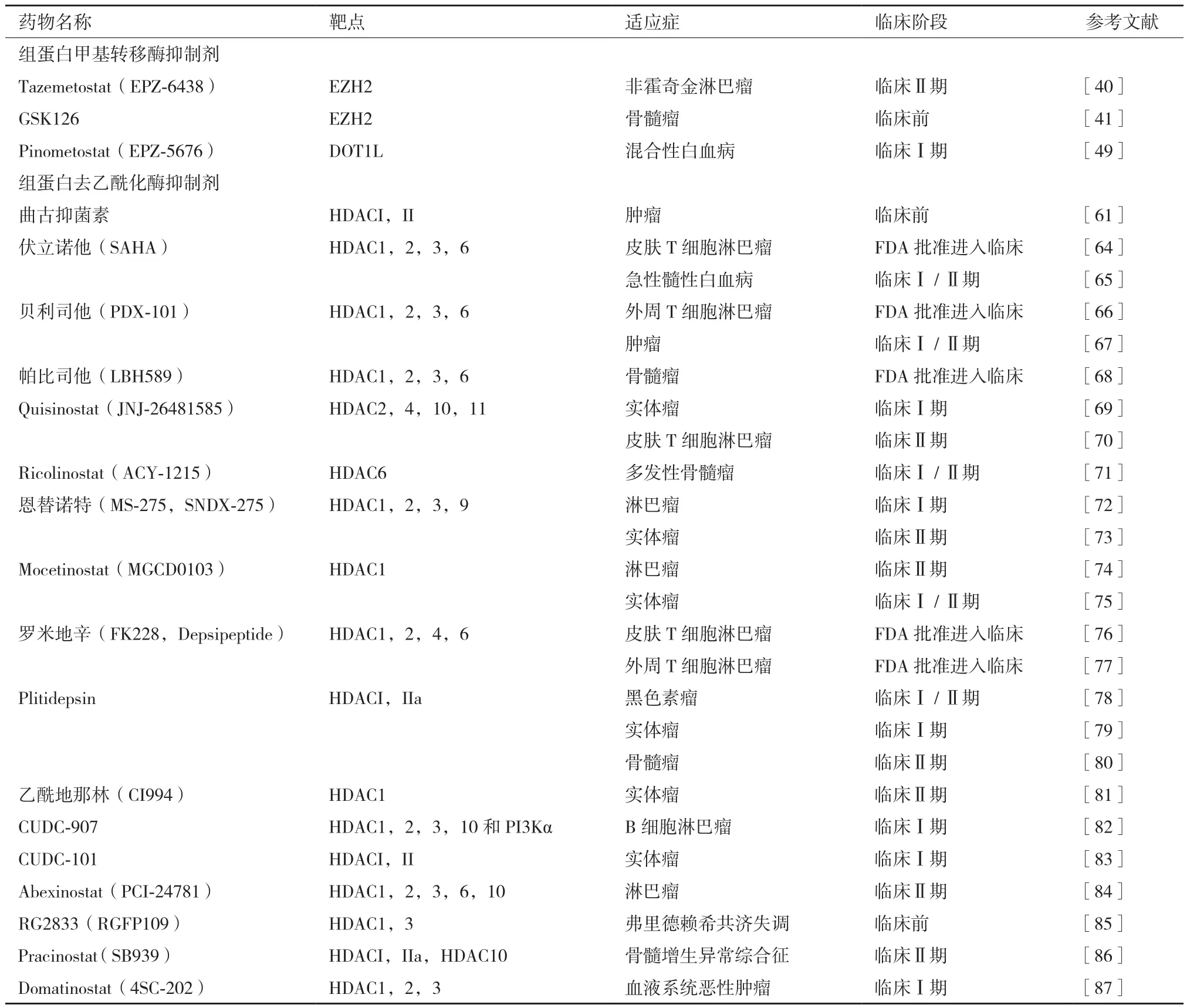
表2 组蛋白修饰酶靶点抑制剂的临床研究阶段
当前,有关HAT抑制剂的研发依然需要面对许多问题,包括很多HAT活性与酶动力学有关的催化机制尚待明确;HAT常常与其他蛋白形成复合体,使得HAT抑制剂在细胞水平和体内疾病模型中的基础研究复杂化。即便如此,借助高通量筛选,已经对大量的化合物的乙酰基转移酶抑制能力进行了筛选,发现一些噻唑类衍生物可降低组蛋白的特异性乙酰化活性,因此噻唑类衍生物可能更具潜力[98]。
2.3 siRNA药物
由于siRNA能特异沉默基因表达,因此可以在肿瘤基因治疗和遗传学上罕见的孤儿疾病的治疗中具有价值。目前,眼部注射、静脉注射及局部递送的siRNA药物研发有了突破性进展。一些siRNA药物表现出良好的临床治疗效果。如PF-655(又名PF-04523655)可靶向沉默RTP801基因,使低氧诱导型因子 1(Hypoxia inducible factor-1,HIF-1)下调,控制细胞增殖和血管发生。在临床I期试验中,将PF-655注射进眼球玻璃体能治疗糖尿病性黄斑水肿(Diabetic macular edema,ME)和年龄相关性黄斑变性(Age-related macular edema,AMD)[99]。除此之外,CALAA-01是用环糊精聚合物包被的siRNA纳米颗粒,可通过降低核糖核苷酸还原酶M2亚基(Ribonucleotide reductase M2 subunit,RRM2) 的 水平抑制肿瘤生长并使肿瘤变小[102]。ALN-VSP02含有两种不同的siRNA分子,用SNALP制成脂质纳米颗粒可分别靶向调控血管内皮生长因子(Vascular endothelial growth factor,VEGF)和纺锤体驱动蛋白(Kinesin spindle protein,KSP/Eg5)[104]。 再 如,Patisiran(又名ALN-TTR01),当用于治疗转甲状腺素蛋白(Transthyretin,TTR)介导的淀粉样变性时,可有效抑制突变型和非突变型转甲状腺素蛋白的产生[106]。ALN-RSV01是针对呼吸道合胞病毒(Respiratory syncytial virus pneumonia,RSV)N-蛋白mRNA研发的抗病毒siRNA药物,在体外和体内表现出特异抑制RSV的活性,可防止感染继发的闭塞性细支气管炎综合征(Bronchiolitis obliterans syndrome,BOS),其雾化剂在临床II期试验中表现了良好的治疗效果(表3)。
当前,siRNA药物处于快速进入临床阶段(表3)[108],但真正大范围应用尚需根本解决以下问题,包括有效降低siRNA药物潜在的“脱靶”风险(在存在碱基错配对的情况下siRNA和靶标mRNA分子发生“互补配对”,并发挥作用);降低siRNA经肾小球过滤快速排出肾脏;避免血管内切酶降解;逃逸溶酶体降解等问题。此外,RNAi可诱发潜在的先天免疫应答[109]。为此,人们尝试在不影响siRNA药效的前提下对siRNA进行化学修饰以提高其抗核酸酶降解能力,增强siRNA的稳定性,或将siRNA包封于脂质体、无机纳米载体或高分子聚合材质的树枝状大分子、聚合物胶束等纳米递送体中以提高siRNA稳定性。很多数据表明,上述纳米递送体既能有效避免siRNA分子在血管内降解,且可降低siRNA分子潜在的免疫毒性,可以极大地改善siRNA药物的药代动力学特征[110]。
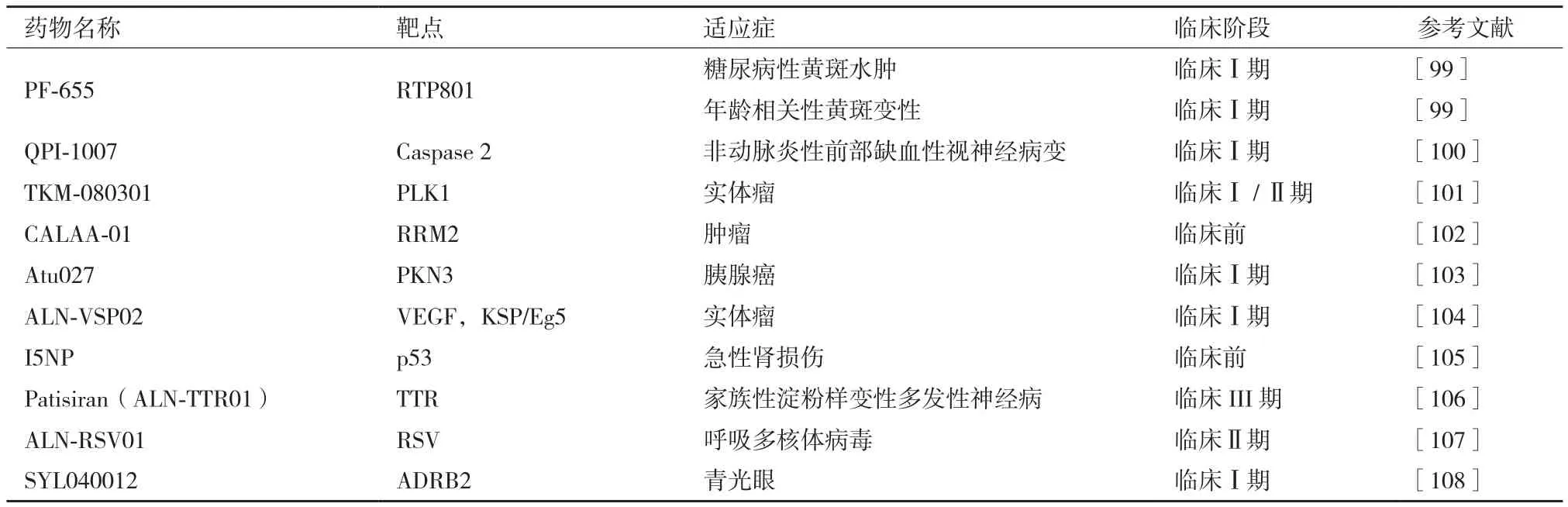
表3 siRNA药物的临床研究阶段
3 展望
染色质表观遗传修饰紊乱可见于肿瘤、糖尿病、心脑血管等诸多重大疾病的病理过程,为此临床上亟需可针对疾病状态下表观遗传修饰紊乱实施干预的药物。多年的临床和实验室研究表明,对表观遗传修饰异常引起的疾病使用表观遗传药物治疗可以更具针对性,疗效明显优于传统抗癌药。在临床实践中,表观遗传药物既可单独使用,也可与传统抗癌药联用,在提高疗效的同时也极大地降低了传统抗癌药的毒副作用。人们普遍期待表观遗传修饰催化剂为靶点的表观遗传药物的发展能为包括肿瘤、糖尿病和心脑血管等众多严重困扰人类健康福祉的重大疾病的根治带来福音。
在取得成效的同时,当前的表观遗传药物的研发也面对着诸多的难题,如siRNA靶向递送药物有潜在的脱靶问题,已上市的阿扎胞苷和地西他滨等干预DNA甲基化修饰的药物能导致骨髓抑制和引发胃肠道症状,而可干预组蛋白修饰的表观遗传药物普遍有细胞毒性等。但是,随着对参与表观修饰催化剂的分子结构和作用机制的深入了解,将不仅有助于从天然产物中筛选有效的表观遗传药物,而且势必对现有基于知识的表观遗传药物的分子设计(Knowledge- based epigenetic drug design)和化学合成起到促进作用,加之包括底物模拟物建模、高通量和虚拟筛选等新方法和新技术的应用,相信表观遗传药物的研发会很快呈现出快速发展的良好态势。
猜你喜欢
分子催化(2022年1期)2022-11-02 07:11:08
中国生物化学与分子生物学报(2022年8期)2022-09-08 00:39:50
保健与生活(2022年11期)2022-06-09 22:24:39
生物学通报(2020年10期)2020-08-13 08:52:24
国际妇产科学杂志(2016年2期)2016-06-16 02:10:22
中国组织化学与细胞化学杂志(2016年4期)2016-02-27 11:15:53
云南中医学院学报(2015年2期)2015-07-31 18:11:59
化学反应工程与工艺(2015年1期)2015-04-16 03:06:16
中国当代医药(2015年16期)2015-03-01 02:03:13
中国塑料(2014年4期)2014-10-17 03:00:50
