
Exploiting autophagy in multiple myeloma
2019-07-31 00:23MatthewHoAshishPatelCathalHanleyAdamMurphyTaraMcSweeneyLiZhangAmandaMcCannPeterGormanGiadaBianchi
Matthew Ho, Ashish Patel, Cathal Hanley, Adam Murphy, Tara McSweeney, Li Zhang, Amanda McCann,2, Peter O’Gorman, Giada Bianchi
1UCD School of Medicine, College of Health and Agricultural Sciences, University College Dublin, Belfield Dublin, Dublin 4,Ireland.
2UCD Conway Institute of Biomolecular and Biomedical Science, Dublin, Dublin 4, Ireland.
3Department of Hematology, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China.
4Haematology Department, Mater Misericordiae University Hospital, Dublin, Dublin 7, Ireland.
5LeBow Institute for Myeloma Therapeutics and Jerome Lipper Multiple Myeloma Center, Department of Medical Oncology,Dana Farber Cancer Institute, Harvard Medical School, Boston, MA 02215, USA.
#These authors contributed equally.
Abstract Multiple myeloma (MM) is a plasma cell cancer characterized by sustained endoplasmic reticulum (ER) stress and unfolded protein response activation in the setting of high rates of immunoglobulin synthesis. Consequently, MM cells rely heavily on protein quality control pathways for survival as evidenced by the clinical efficacy of proteasome inhibitors (PI). Autophagy is an intracellular self-digestion mechanism that plays a role in the ER protein quality control process. Unsurprisingly then, basal levels of autophagy were recently found to confer a survival and drugresistance benefit to MM cells. However, excessive induction of autophagy in MM cells leads to autophagic cell death, highlighting the double-edged nature of autophagy modulation in MM. This review provides an overview of the role that autophagy plays in MM pathogenesis, survival, and drug-resistance. We highlight the potential utility of therapeutically targeting autophagy in MM, focusing on preclinical data of autophagic modulators in combination with alkylators, anthracyclines, PI, and immunomodulatory drugs.
Keywords: Multiple myeloma, autophagy, drug resistance, hematopoiesis, immunoglobulin, antibody
MULTIPLE MYELOMA
Multiple myeloma (MM) is a neoplastic disorder characterized by the dysregulated proliferation of a plasma cell clone that typically produces a monoclonal immunoglobulin, ultimately resulting in end-organ damage[1-3]. Clinical suspicion for active MM is often based on the presence of one or more laboratory/imaging abnormalities, termed the CRAB criteria [hypercalcaemia (C), renal impairment (R), anaemia (A),and osteolytic bone lesions (B)], particularly if occurring in a patient with a precursor plasma cell disorder such as monoclonal gammopathy of undetermined significance (MGUS) or smoldering MM (SMM)[4]. A diagnosis of active MM requires the presence of greater than 10% clonal bone marrow (BM) plasma cells in association with either one or more of the CRAB features or a biomarker of malignancy (BM plasmacytosis equal or greater than 60%, ratio of involvedvs. uninvolved light chain equal or greater than 100 or the presence of more than 1 focal lesion on magnetic resonance imaging)[5,6]. MM is generally preceded by the asymptomatic precursor conditions MGUS and/or SMM[6]. MGUS is characterized by low levels of monoclonal protein (< 3 g/dL) and less than 10% clonal plasma cells in the BM while SMM is characterized by the presence of > 3 g/dL of monoclonal protein with BM plasmacytosis exceeding 10% but less than 60%[6]. Evidence of end organ damage related to the plasma cell disorder is an exclusion criteria for MGUS/SMM diagnosis. Patients with MGUS and SMM progress to active MM at a rate of 1% and 10% per year,respectively[7].
While single driver mutations have not been identified in MM, marked genomic instability is a hallmark of the disease and contributes to elevated proteotoxic stress[8]. The high frequency of genomic mutations may confer a survival advantage by enabling MM cells to quickly adapt to stresses in the environment.However, this comes at a cost. This deregulation of gene expression results in the accumulation of toxic misfolded proteins that exerts additional stress on MM cells[9-13]. Furthermore, MM are highly secretory cells, characterized by staggering rate of synthesis of clonal immunoglobulins which further contributes to baseline ER stress. Therefore, protein quality control pathways are essential for MM survival[8].
AUTOPHAGY
Autophagy is a tightly regulated self-digestion mechanism that promotes the lysosomal degradation of organelles, intracellular pathogens, and misfolded proteins. It is a key cellular mechanism to maintain homeostasis and guarantee energy supply as products of autophagic digestion can be re-utilized in anabolic processes[14-16]. Therefore, nutrient and energy deprivation, ER stress, and hypoxia can all induce autophagy as a means to enable cell survival[17]. In mammalian cells, there are three main types of autophagy, namely macroautophagy, microautophagy, and chaperone-mediated autophagy[15].
Macroautophagy
Macroautophagy is a type of autophagy that delivers cellular contents to the lysosome via the formation of double-membrane structures called autophagosomes which then fuse with lysosomes to form autolysosomes[18,19]. Macroautophagy can be subdivided into non-selective (bulk) and selective autophagy[16].During non-selective autophagy, bulk cytoplasm is randomly engulfed by a phagophore [Figure 1]. Notably,the mammalian target of rapamycin (mTOR) pathway is a key inhibitor of autophagy[20]. Subsequently,the phagophore matures into an autophagosome and this process is mediated by autophagy-related protein 7(ATG7), ATG8 (LC3), and ATG12[21,22]. ATG7 functions as an E1-like enzyme by binding and activating ATG12 and ATG8 to facilitate the transfer of ATG12 to ATG5 via the E2 enzyme ATG10[23-27]. The resultant ATG12-ATG5 conjugate forms a large multimeric complex together with ATG16 (ATG12-ATG5-ATG16) which acts as an E3 ligase to facilitate phosphatidylethanolamine (PE) and LC3 conjugation and conversion of LC3-I to LC3-II. LC3-II stably associates with the autophagosome membrane and regulates autophagic membrane expansion, recognition of autophagic cargo, and autolysosome formation[21,22]. Finally, autophagosome and lysosome fusion occurs and the autophagic cargo is degraded by lysosomal hydrolases[21,22]. Selective macroautophagy, on the other hand, specifically targets damaged or redundant organelles such as mitochondria (mitophagy), peroxisomes (pexophagy), ribosomes (ribophagy), aggresomes (aggrephagy),etc.[28]. Specifically, mitophagy is the selective degradation of mitochondria by macroautophagy in a PTEN-induced kinase 1 (PINK1)- and Parkin-dependent fashion[29,30]. Type 1 mitophagy sequesters and removes mitochondria in response to nutrient deprivation, whereas type 2 mitophagy removes damaged mitochondria[31]. Type 3 mitophagy (micromitophagy), on the other hand, eradicates damaged mitochondrial components through the formation of mitochondria-derived vesicles that are subsequently degraded by lysosomes[31].
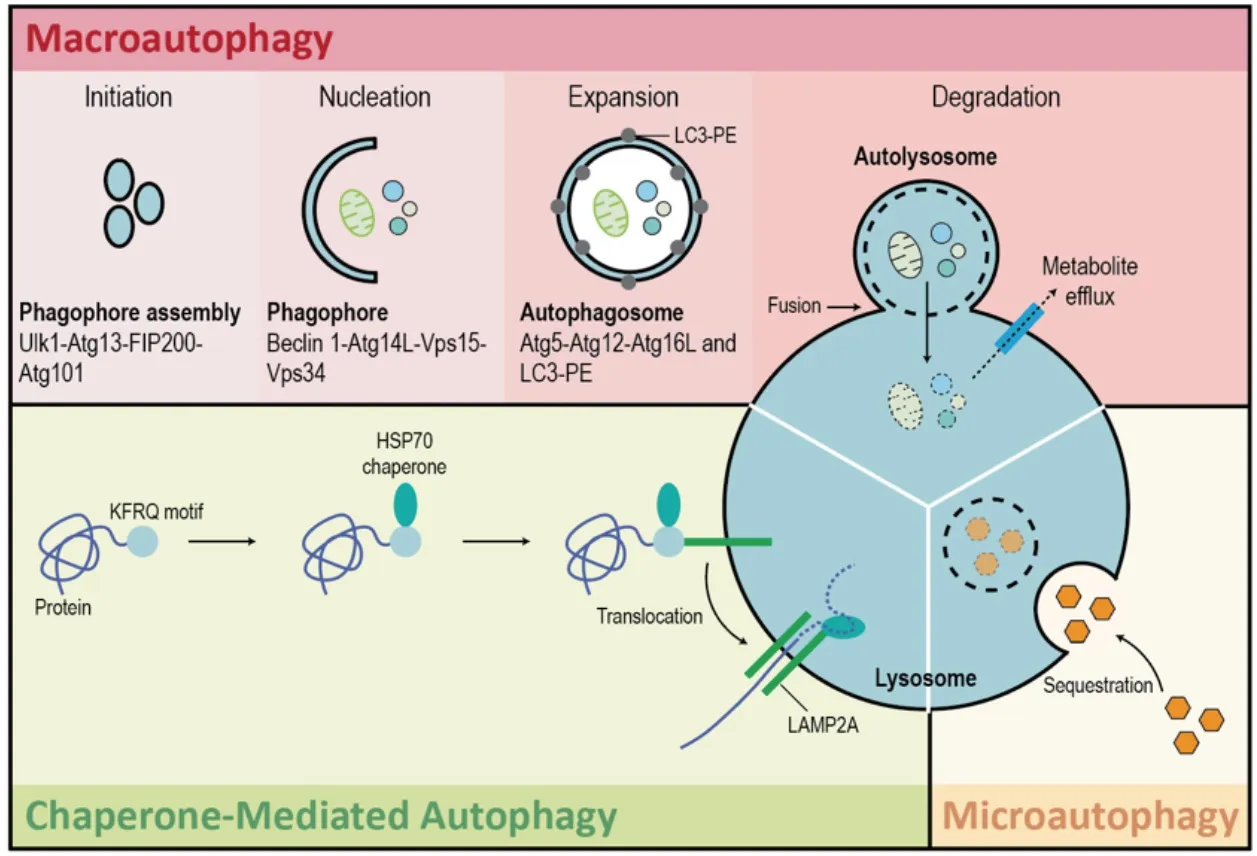
Figure 1. Autophagy. There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy.Macroautophagy is a type of autophagy that delivers cellular contents to the lysosome via the formation of double-membrane structures called autophagosomes which then fuse with lysosomes to form autolysosomes. Macroautophagy takes place in five main steps. Initiation of autophagy occurs in response to metabolic or therapeutic stress and is mediated by ULK1, ATG13, FIP200, and ATG101. During the nucleation step regulated by BECLIN-1, ATG14L, VPS15, and VPS34, the formation of the phagophore occurs. Expansion results in the sequestration of cytosolic contents within the autophagosome and is facilitated by ATG5, ATG12, ATG16L, and LC3-PE. Degradation is the breakdown of autophagosomal contents upon formation of the autolysosome (fusion of autophagosome and lysosome). Microautophagy is a largely non-selective process that facilitates the direct uptake and breakdown of cytosolic cargo by lysosomes. Chaperone-mediated autophagy refers to the chaperone-dependent targeting of specific cytosolic proteins to lysosomes for proteolysis. HSC70 binds to the consensus motif of specific proteins to target them to the lysosome-associated membrane protein type 2A (LAMP-2A) receptor on the lysosomal membrane. Once internalized by the lysosome, these cytosolic proteins are degraded
Microautophagy
In eukaryotic cells, microautophagy is a largely non-selective process that facilitates the direct uptake and breakdown of cytosolic cargo by lysosomes. Specifically, cytosolic material is sequestered by direct invagination of the vacuolar/lysosomal membrane, forming autophagic tubes that pinch off into the lysosomal lumen[32-34].
Chaperone-mediated autophagy
Chaperone-mediated autophagy (CMA) refers to the chaperone-dependent targeting of specific cytosolic proteins to lysosomes for proteolysis[35-37]. This is a mechanistically distinct process that occurs only in mammalian cells. Unlike the other types of autophagy, CMA does not require the formation of vesicles[37].Instead, HSC70 binds to the consensus motif of specific proteins to target them to the lysosome-associated membrane protein type 2A receptor on the lysosomal membrane[35-37]. Once bound, the targeted proteins start to unfold as they are internalized into the lysosomal lumen and then degraded[35-37].
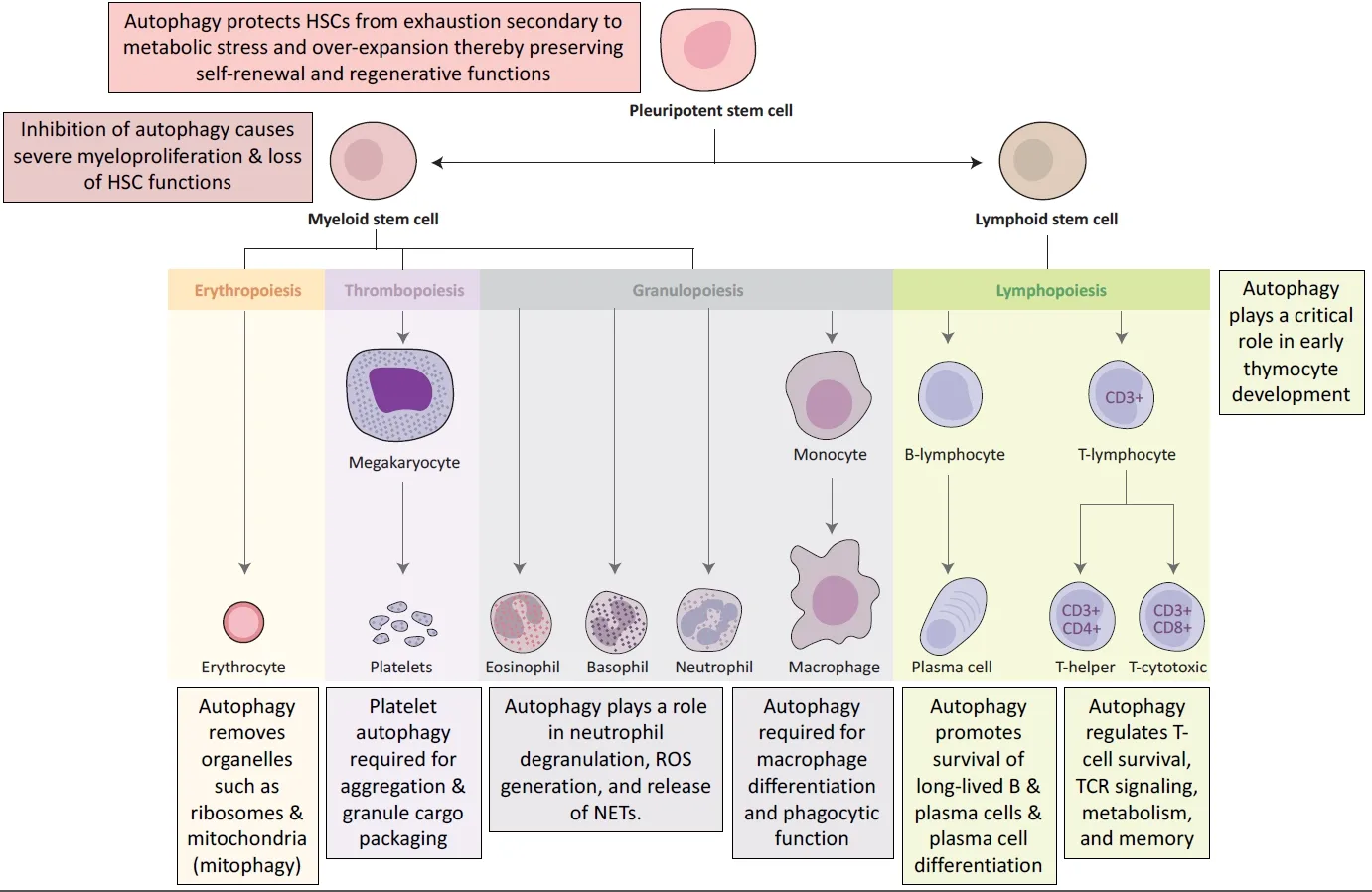
Figure 2. Examples of key functions that autophagy plays in the maintenance and differentiation of hematopoietic stem cells. Image adapted from First Aid for the USMLE Step 1[63]
IMMUNE FUNCTIONS OF AUTOPHAGY
Autophagy as an immune effector
Autophagy plays a key role in eukaryotic cells as a stress response mechanism activated to recycle intracellular organelles in the face of nutrient deprivation[38]. However, advances in our understanding of autophagy reveal also an intricate reciprocal relationship between autophagy and immunity[38]. Specifically,autophagy acts as an immune effector by: (1) facilitating the direct elimination of microbes through xenophagy and LC3-associated phagocytosis[39]; (2) modulating the inflammatory response by amplifying PAMP-TLR signaling whilst inhibiting type I IFN signaling and inflammasome activation[40]; (3) enhancing MHC class II-mediated antigen presentation and cross-presentation[39,40]; and (4) contributing to secretion of pro-inflammatory cytokines such as IL-6, which could be relevant to MM pathogenesis[39].
Autophagy in the maintenance of hematopoiesis
Apart from regulating immune effector functions, autophagy is a key adaptive mechanism critical to the maintenance of hematopoietic hemostasis [Figure 2][41]. Hematopoietic stem cells (HSCs) thread a tight,but dynamic and demand-adapted, balance between quiescence and proliferation throughout the entire life of the organism[41]. Studies have shown that autophagy protects HSCs from exhaustion secondary to metabolic stress[41-43]. Firstly, basal autophagy removes activated mitochondria, regulates the metabolism,and maintains the self-renewal and regenerative capabilities of HSCs[44]. Research shows that conditional deletion of ATG12 in transplanted murine HSCs severely impairs their ability for BM engraftment and selfrenewal[45]. Secondly, when subjected toex vivocytokine withdrawal orin vivonutrient deprivation, HSCs robustly upregulate autophagy to circumvent an energy crisis to ensure HSC longevity[43].
Deletion of ATG7, a critical component of autophagosome formation, in murine HSCs results in accumulation of dysfunctional mitochondria, upregulation of oxidative stress and DNA damage, and ultimately cell death[46]. Interestingly, aged HSCs were found to maintain a low metabolic state by upregulating autophagy in order to sustain robust long-term self-renewal potential comparable to young HSCs[44]. Downstream of HSCs, autophagy also plays a key role in the development, differentiation, and function of erythrocytes, platelets, granulocytes, macrophages, and T cells as summarized in Figure 2[41].
Since malignant transformation of HSCs or early progenitors results in leukemia, homeostatic mechanisms;such as autophagy, that protect HSCs from metabolic, oxidative, and genotoxic stress; are crucial to prevent hematopoietic malignancies[47]. Indeed, ATG7 deletion in myeloid cells results in dysregulated and invasive myeloproliferation resembling acute myeloid leukemia[42].
Autophagy in plasma cell ontogeny
Autophagy plays a key role in plasma cell (1) differentiation; (2) survival; and (3) protein quality control.
Autophagy in plasma cell differentiation and survival
B lymphocyte to plasma cell differentiation is controlled by a complex genetic reprogramming system leading to downregulation of genes involved in the maintenance of B-cell identity [e.g., paired box protein 5 (PAX5),transcription regulator protein BACH2 (BACH2), B-cell lymphoma protein 6 (BCL6)] and upregulation of genes involved in terminal differentiation of Ig-secretory plasma cells [e.g., B-lymphocyte-induced maturation protein 1 (BLIMP-1), interferon regulatory factor 4 (IRF4), and X-box binding protein 1 (XBP1)][48].Specifically, BLIMP-1 acts as a molecular switch to repress PAX5 and BCL6, and induces XBP1 to promote antibody production and plasma cell differentiation[49,50]. Interestingly, while BLIMP-1 and IRF4 are essential for plasma cell differentiation, only IRF4 is essential for plasma cell survival[51]. Consistent with this, BLIMP-1 deficient plasma cells remained viable and retained their transcriptional identity but lose the ability to secrete Ig[51].
Beyond epigenetics, autophagy also plays an essential role in plasma cell differentiation and survival[Figure 3]. Studies have found increased expression of autophagic genes in differentiating plasma cells.Conditional deletion of ATG5 in murine B cells results in reduced IgM and IgG responses in the setting of both T-cell dependent and independent immunizations; further suggesting that autophagy is required for B lymphocyte to plasma cell differentiation[52]. Notably, ATG5 was also essential for the homing and/or survival of long-lived plasma cells in the BM[52]. Consistent with this, long-lived plasma cells were found to highly express autophagic genes and display high basal levels of autophagy[53].
Autophagy as a mechanism of protein quality control
Plasma cells are professional antibody secreting cells (ASCs) uniquely optimized towards large-scale immunoglobulin synthesis, folding, assembly, and secretion[54]. Not unique to plasma cells, however, is the fact that the protein synthesis process is intrinsically error prone. In fact, up to 30% of newly-synthesized proteins are defective and need to be degraded[47,54]. Thus, an intricate balance between protein synthesis,folding, and clearance must be maintained to prevent the accumulation of potentially toxic misfolded proteins[47,54]. This is especially crucial for ASCs that cope with increased Ig synthesis by upregulating folding capacity through the induction of unfolded protein response (UPR)-driven ER expansion[11].However, when Ig synthesis exceeds folding capacity, the integrity of the proteome is preserved through an interconnected network of protein quality control pathways which include the proteasome, autophagy,aggresome, and UPR pathways[47]. These pathways are so important to plasma cells that the amount of newly-synthesized proteins degraded by the proteasome is 15-folds higher in plasma cells compared to resting B-cells[55]. Perhaps not surprisingly then, that MM, a cancer of plasma cells, exhibits the same reliance on the protein quality control as evidenced by the clinical efficacy of proteasome inhibitors (PI).
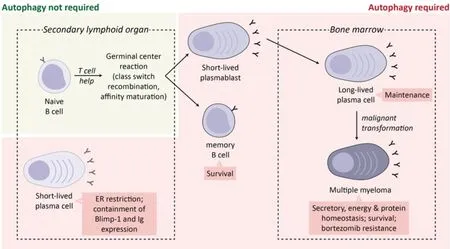
Figure 3. Role of autophagy in plasma cell ontogeny and malignancy. Differentiating plasma cells and MM cells induce autophagy to restrict the ER and downregulate BLIMP-1 expression to decrease immunoglobulin synthesis and deal with excessive proteotoxic stress.Autophagy is also essential for the survival and maintenance of memory B cells and BM long-lived plasma cells. In MM, induction of autophagy has been linked with bortezomib resistance. Image adapted from Milan et al.[60]
Indeed, sensitivity of MM to PI is determined by a combination of Ig secretory load, protein degradation capacity, and commitment to plasma cell maturation; which, taken together, suggests that being an ASC confers this unique susceptibility to MM[56-59].
While autophagy does not directly dispose of misfolded immunoglobulins, plasma cells deficient in autophagy had greater energy imbalance, enhanced immunoglobulin synthesis, reduced intracellular ATP, and elevated ER stress[52]. Consistent with this, autophagy-deficient plasma cells displayed higher expression of BLIMP-1 and XBP1 with corresponding increase in ER size [Figure 3][52]. These findings suggest that autophagy not only facilitates the acquisition of an Ig-secretory phenotype in the in preplasma cells, but also serves to limit the overproduction of Ig and maintain cellular ATP levels to ensure survival in differentiated plasma cells [Figure 3][60]. Autophagy also plays an important role in the removal of misfolded protein aggregates which, due to steric hindrance, are unable to be efficiently degraded by the proteasome[61]. Failure to dispose of misfolded protein aggregates results in proteotoxic stress and induction of terminal ER stress[62].
AUTOPHAGY IN MM
Autophagy is necessary for the differentiation and maintenance of antigen-specific long-lived plasma cells,the physiologic counterpart to MM cells. As MM cells retain many characteristics of the original plasma cell clone such as Ig secretion and an enlarged cytoplasm and ER, it is reasonable to hypothesize that autophagy might also play a role in MM proteostasis and survival.
Indeed, MM cells exhibit higher levels of basal autophagy compared to other tumors, including lymphoma derived from earlier B-cell progenitors, and autophagy is necessary for the survival of MM cells[64]. Disruption of autophagy through BECLIN-1 knockdown or pharmacologic inhibition (with 3-methyladenine and/or chloroquine) causes MM cell apoptosis[65,66]. Basal autophagy is hypothesized to alleviate proteotoxic stress and promote MM survival by (1) limiting the secretion of immunoglobulins and (2) providing an alternative proteolytic pathway for the clearance of ubiquitinated proteins through a p62-dependent mechanism[60]. Despite this, concomitant inhibition of the proteasome and autophagy in pre-clinical studies have reported inconsistent results, ranging from synergism to antagonism[60]. This may partly be explained by the observation that while basal autophagy is protective, persistent and uncontrolled autophagy results in autophagic cell death[67,68]. Autophagic cell death in MM can be induced by caspase-10 or caspase-8 inhibition[67,68]. Specifically, caspase-10 cleaves and inhibits BCL2 associated transcription factor 1, a BECLIN-1 activator, to temper the autophagic response and avoid cell death[67]. MM cells therefore need to tightly regulate autophagy to maintain viability as dysregulation either way leads to deleterious effects in MM.
Mitophagy has been reported to play conflicting roles in carcinogenesis, survival, and drug-resistance.Some studies report that mitophagy protects untransformed cells from excessive reactive oxidative species damage and genetic instability, and that suppression of mitophagy favors carcinogenesis[69,70]. Conversely,other studies suggest that mitophagy promotes cancer cell survival and drug resistance by protecting cells from apoptosis[71,72]. In MM, while the suppression of mitophagy is associated with bortezomib resistance;doxorubicin, a widely used anti-MM chemotherapy, is a classical inhibitor of mitophagy[73]. This dichotomy highlights the need for a better understanding of the role mitophagy plays in MM.
DNA damage and autophagy
Apart from ongoing proteotoxic stress, other hallmarks of MM are genetic instability and abnormalities(e.g., aneuploidy, translocations), and oxidative stress which lead to replicative stress and constitutive DNA damage[74,75]. Recent studies have shown that DNA damage can activate autophagy through a number of interconnected pathways[76]. Examples of DNA damage repair (DDR) pathway proteins that regulate autophagy are ataxia telangiectasia-mutated (ATM), poly (ADP-ribose) polymerase 1 (PARP1),c-Jun N-terminal kinase (JNK), and p53[76]. Specifically, autophagy can be induced by ATM-mediated phosphorylation of AMP-activated protein kinase (AMPK)[77,78]. AMPK in turn activates both tuberous sclerosis complex 2 (TSC2), to remove the inhibitory effect of MTOR complex 1 (mTORC1) on autophagy,and unc-51 like autophagy activating kinase 1 (ULK1) to promote autophagosome formation[77-79]. ATM also phosphorylates and activates CHE-1 which in turn upregulates the transcription of two mTOR inhibitor genes [regulated in development and DNA damage responses 1 (REDD1) and DEP domaincontaining mTOR-interacting protein (DEPTOR)][80]. Nuclear factor-kappa B, a well-known pro-myeloma transcription factor, can be activated by ATM and as a result, upregulate BECLIN-1[81]. Induction of PARP1 promotes autophagy via AMPK activation on the background of ATP depletion and elevated AMP levels[82].DNA damage-induced JNK phosphorylates BCL-2 resulting in BCL-2 dissociation, relief of BECLIN-1 inhibition, and induction of autophagy[83]. Nuclear p53 activates autophagy through a number of distinct signaling pathways. Firstly, p53 upregulates phosphatase and tensin homolog (PTEN) which leads to PI3KAkt-mTORC1 inhibition and autophagy induction[84,85]. Secondly, p53 influences AMPK activity (1) directly through transcriptional upregulation, and (2) indirectly by activating Sestrin1 and Sestrin2 which in turn activate AMPK[85,86]. Thirdly, death-associated protein kinase (DAPK) is transcriptionally upregulated by p53 and triggers autophagy by phosphorylating BECLIN-1 to facilitate its dissociation from BCL-2 and BCL-XL[87-89]. DAPK also phosphorylates protein kinase D which activates the VPS34 class III PI3K complex leading to induction of autophagy[87-89]. Lastly, p53 also upregulates damage-regulated autophagy modulator, a lysosomal protein involved in the degradation step of autophagy[90]. However, unlike nuclear p53, cytoplasmic p53 can also activate mTOR to inhibit autophagy[91]. Functionally, autophagy is essential for homologous recombination and nucleotide excision repair and cells deficient in autophagy rely chiefly on the error-prone non-homologous end joining repair pathway, which may explain the genomic instability observed in autophagy-deficient cells[92-98].
EXPLOITING AUTOPHAGY IN MM
Preclinical studies of autophagy modulators in MM
HDAC6 inhibitors
Histone deacetylases (HDACs) catalyze the removal of acetyl groups on lysine residues in target proteins[99].Furthermore, HDACs also deacetylate non-histone proteins, thereby providing an additional layer of control over protein function, stability, and protein-protein interaction[99]. HDAC6 is a class IIb HDAC that is mainly localized to the cytoplasm, unlike class I and IIa HDACs which shuttle between the cytoplasm and nucleus, suggesting that HDAC6 functions mainly to regulate non-histone proteins[99]. Indeed,HDAC6 possess intrinsic ubiquitin-binding activity and co-localizes with the microtubule network to transport misfolded polyubiquitinated proteins to aggresomes/autophagosomes for subsequent lysosomal degradation[99-101]. HDAC6 therefore promotes ubiquitin-dependent or ubiquitin-independent aggresome formation and while HDAC6 is not required for the initiation of autophagy per se, it is necessary for the targeted delivery of the autophagy machinery to aggresomes[62,100-105]. Importantly, HDAC6 also promotes the formation of an F-actin network essential for autophagosome-lysosome fusion[105]. The development of HDAC6 inhibitors therefore presents an opportunity to exploit autophagy to destabilize protein homeostasis in MM.
Preclinical studies have shown that the HDAC6-selective inhibitors WT161 and Tubacin trigger the accumulation of acetylated tubulin of and inhibits MM cell growthin vitro[106,107]. Additionally, combinatory treatment using WT161 or Tubacin with the PI bortezomib not only induces synergistic cytotoxicity but was also able to overcome bortezomib resistance[106,107]. Mechanistically, the addition of WT161 to bortezomib resulted in further accumulation of polyubiquitinated proteins which led to a further increase in ER stress signaling and the UPR, evidenced by the upregulation of ATF4 and the pro-apoptotic protein CHOP[106]. Interestingly, the ER stress sensor proteins inositol-requiring enzyme 1 (IRE1α) and PRKR-like endoplasmic reticulum kinase (PERK) were found to be downregulated by WT161, suggesting that HDAC6 inhibition also suppresses the UPR, preventing it from functioning as an ER stress mitigator[106].
Another HDAC6-selective inhibitor, ACY-1215, demonstrated synergistic cytotoxicity when used in combination with carfilzomib, an irreversible second generation PI[108]. The addition of ACY-1215 resulted in increased LC3-II consistent with the disruption of autophagic flux secondary to HDAC6 inhibition[108].Mechanistically, the addition of ACY-1215 to carfilzomib inhibited both aggresome formation and autophagosome-aggresome association[108]. Consistently, a novel HDAC6 inhibitor MPT0G413 was shown to both disrupt bortezomib-induced aggresome formation and exhibit synergism with bortezomib[109].The combination of MPT0G413 and bortezomib could also overcome cell adhesion-mediated drug resistance in the MM-BMSC co-culture setting[109]. MPT0G413 was further found to decrease MM-BMSC adhesion and downregulate the pro-myeloma cytokines VEGF and IL-6 in the context of the MM BM microenvironment[109].
Autophagy inhibitors + DNA-damaging chemotherapy
DNA damaging agents such as melphalan are highly cytotoxic to MM cells and are a mainstay of treatment in MM, particularly as a conditioning regimen ahead of autologous hematopoietic stem-cell transplantation[110]. Consistent with the aforementioned link between DDR and autophagy, a recent study found that melphalan and doxorubicin induce cytoprotective autophagy as a pro-survival mechanism in MM cells through the upregulation of BECLIN-1-dependent autophagosomes[111]. knockdown of autophagy genes (BECLIN-1 and ATG5) and pharmacologic inhibition of autophagy (via hydroxychloroquine and 3-methyladenine) significantly enhanced the cytotoxicity of melphalan and doxorubicinin vitroandin vivo[111].
High mobility group protein B1 (HMGB1) is a critical regulator of autophagy that is often upregulated in MM[112,113]. Specifically, HMGB1 binds competitively to BECLIN-1 causing BCL-2 displacement and autophagy induction[112]. A study found that HMGB1 overexpression in MM was associated with mTOR inhibition, autophagy induction, and reduced sensitivity to dexamethasone[113]. Conversely, knockdown of HMGB1 increased apoptosis in MM cells exposed to dexamethasone[113].
HMGB1 has also been recognized a marker of induction of immunogenic cell death (ICD). ICD is triggered by exposure to certain cytotoxic agents (e.g., anthracyclines, oxaliplatin, bortezomib)[114,115]. ICD involves the release of soluble mediators along with alterations in the cancer cell surface composition in a way that converts dying cancer cells into therapeutic vaccines that are phagocytosed by antigen presenting cells(APCs) for the cross-priming of CD8+ T-cells to stimulate anti-tumor specific T-cell immune responses[114,115].HMGB1 is a nuclear nonhistone chromatin-binding protein secreted by dying tumor cells exposed to cytotoxic agents that binds to toll-like receptor 4 on APCs to increase the rate of antigen processing and presentation by APCs to T cells[116]. Interestingly, studies have shown that autophagy in tumor cells regulates HMGB1 secretion and that inhibition of autophagy leads to intracellular sequestration of HMGB1 and the induction of caspase-mediated cell death[117]. Macroautophagy therefore enhances antigen crosspriming after ICD which results in the following conundrum. From a cytotoxicity point of view, it makes sense to combine DNA-damaging agents with autophagy inhibitors. However, from an ICD perspective,the addition of autophagy inhibitors to DNA-damaging agents may be counterproductive to antigen crosspriming.
Autophagy modulators + PI
Bortezomib, a first-in-class PI, was approved for use in MM by the FDA in 2003 and has become a mainstay of therapy at every disease stage ever since. Despite significant clinical efficacy in most naïve patients, most of the patients ultimately become refractory to bortezomib therapy[118-120]. This has prompted the development of second-generation PIs (e.g., carfilzomib and ixazomib) currently in clinical use for relapsed or refractory MM; and next-generation PIs (e.g., marizomib and oprozomib) currently in advanced clinical trials[121]. Based on evidence that susceptibility to PI depends on the ability of MM cells to remove misfolded proteins through protein quality control pathways, novel therapeutic approaches targeting alternative pathways in protein homeostasis are also actively being studied[122].
Research has shown that crosstalk exists between the proteasome, UPR, and autophagy[65,123,124]. Specifically,whenever the proteasome is overwhelmed and/or inhibited, polyubiquitinated and misfolded proteins coaggregate in the cytosol to form aggresomes which are then degraded through macroautophagy[101,125,126].Mechanistically, proteasome inhibition induces ER stress and PERK-eIF2α and IRE1-JNK activation,leading to the induction of autophagy[127,128]. Other mechanisms of bortezomib resistance, in the context of autophagy, include the upregulation of PROFILIN-1 which enhances autophagy through BECLIN-1 interaction and increased expression of CIC5, a chloride channel that enhances bortezomib-induced autophagy via AKT-mTOR inhibition[129]. PIs also rapidly induce the expression of SQSMT1/p62 to upregulate p62-dependent autophagy to compensate for proteasome insufficiency[64].
Along the same line, preclinical studies have reported synergistic cytotoxicity with dual blockade of the proteasome and autophagy in MM. MG132, a PI, induced cytoprotective autophagy that can be inhibited,by 3-methyladenine, to enhance apoptotic cell death[128]. Notably, 3-methyladenine resulted in the accumulation of polyubiquitinated proteins and exacerbation of ER stress in cells treated with MG132[128].Autophagy inhibitors that have demonstrated synergy and enhanced MM cytotoxicity with bortezomibin vitroinclude Elaiphyllin (macrolide antibiotic) and Metformin[130,131]. Elaiphyllin, in particular, had significant cytotoxic activity even when used alone in mutant p53 MM[130]. Metformin, on the other hand,inhibits GRP78 which is crucial mediator of bortezomib-induced protective autophagy[131].
However, other conflicting studies have also shown that Metformin inhibits MM proliferation by inducing autophagy (and cell cycle arrest) through AMPK activation and dual repression of mTORC1 and mTORC2[132,133]. In line with this, it has been reported that when used in combination with bortezomib,the autophagy inhibitors 3-methyladenine or chloroquine can also have an antagonistic effect[65]. One potential explanation could be that bortezomib induces apoptosis partially through autophagic cell death.Consistent with a role for autophagic cell death in MM cells treated with bortezomib, a novel SCF (Skp2)inhibitor CpdA stabilizes p27 to induce caspase-independent autophagic cell death in MM cells resistant to bortezomib; and also synergized with bortezomib[134]. Another compound, betulinic acid (BetA), activates protein phosphatase 2A (PP2A) to trigger DAPK-dependent autophagic cell death in MM cells with high BCL-2 expression[135].
Autophagy inhibitors have also shown potency in bortezomib and carfilzomib-resistant MM cells[136]. The non-selective HDAC inhibitor SBHA upregulates the BH3-only protein BIM, which in turn sequesters BECLIN-1 to inhibit cytoprotective autophagy, thereby overcoming acquired bortezomib resistance[136].Chloroquine potentiated carfilzomib cytotoxicity and was able to overcome carfilzomib resistancein vitro[137]. Lastly, pharmacologic inhibition of thioredoxin with PX12 upregulates mitophagy to resensitize bortezomib-resistant cells to bortezomib[73]. Combination treatment of MM cells with PX12 and bortezomib led to synergistic toxicity and inhibition of ERK1/2 and mTOR signaling, suggesting the involvement of ERK1/2 and mTOR in mitophagy suppression[73].
PI3K-AKT-mTOR inhibitors
While activating mutations in PI3K and AKT have not been reported in MM, the PI3K-AKT-mTOR pathway is commonly activated in MM through juxtacrine and paracrine signaling within the MM tumor microenvironment[138,139]. Interactions between MM and the stromal and endothelial compartments result in the secretion of IL-6, VEGF, and IGF-1, which in turn activate pro-survival and proliferative pathways such as PI3K-AKT-mTOR, JAK/STAT3, NFkB, and MEK/ERK[139]. In line with this, efforts to inhibit PI3KAKT-mTOR signaling have led to the study of mTORC1 inhibitors (e.g., rapamycin) in MM. However,results from preclinical studies were disappointing as rapamycin and the other rapalogs demonstrated a cytostatic, but not cytotoxic, response in MM[140]. This lack of potency could be due, in part, to the activation of cytoprotective autophagy. Nonetheless, mTORC1 inhibitors have shown synergistic activity in other cancers when used in combination with clinically approved anti-MM agents such as dexamethasone,lenalidomide, and panobinostat, and pre-clinical agents such as sorafenib (tyrosine kinase inhibitor),17-AAG (HSP90 inhibitor), NVP-AEW541 (IGF1R inhibitor), and MK2206 (AKT inhibitor)[141-148].
Upstream of mTOR, AKT inhibitors have also been studied in MM. In preclinical studies, perifosine,an AKT inhibitor, was cytotoxic to MM cells as a single-agent and also synergized with bortezomib,dexamethasone, doxorubicin, melphalan, and U0126 (MEK1/2 inhibitor)[149]. Another AKT inhibitor TAS-117 enhanced ER stress and MM apoptosis when added to bortezomib or carfilzomib[150]. These studies once again highlight the duality of autophagy in MM, suggesting that excessive induction of autophagy, rather than protecting cells from excessive ER stress, pushes cells towards autophagic cell death.
The next class of PI3K-AKT-mTOR pathway inhibitors are the dual PI3K-mTOR inhibitors. One such inhibitor, BEZ235, demonstrated good preclinical single-agent activity in MM and also synergized with bortezomib, doxorubicin, and melphalan[151,152]. Another study reported a pro-survival function of autophagy in MM cells treated with PI-103, a competitive dual PI3K and mTOR inhibitor, which inhibits the proteasome, induces UPR, and upregulates autophagy[153]. Importantly, Bafilomycin-A, an autophagy inhibitor, enhanced apoptosis in cells treated with PI-103[153].
Heat-shock protein inhibitors
Heat shock proteins are molecular chaperones that play indispensable roles in protein folding/unfolding,multiprotein complex assembly, and protein sorting[154]. As a function of protein sorting, HSP70 and HSP90 also participate in chaperone-mediated autophagy[155-157]. Heat-shock proteins (HSPs) therefore help alleviate proteotoxic stress to prevent apoptosis in MM[158]. Consistent with this, HSP70 and/or HSP90 inhibition induces UPR and apoptosis in MM[159-161]. In preclinical studies, combination of HSP90 inhibitors KW-2478,Retaspimycin, and 17-AAG with bortezomib demonstrated synergistic cytotoxicity[162-164]. Another HSP90 inhibitor, NVP-HSP990, displayed potent,in vitroanti-myeloma activity and synergism with melphalan,HDAC inhibitors, and PI3K/mTOR inhibitors[165,166]. Other HSP90 inhibitors such as PU-H71, SNX5422,and NVP-AUY922 have also shown promising pre-clinical results in MM[158,167-169]. Apart from HSP90,HSP70 has recently emerged as a promising therapeutic target in MM and a number of HSP70 inhibitors(e.g., PET-16, Ver-155008, MAL3-101) have shown good pre-clinical anti-myeloma activity[160,161,170,171].
Interestingly, treatment of MM cells with HSP90 inhibitors (e.g., 17-AAG, NVP-AUY922), bortezomib, or dexamethasone results in compensatory upregulation of HSP70 which confers a degree of drug-resistance and protects MM cells from apoptosis[172,173]. HSP70 is a molecular chaperone of HSP90. Consequently,inhibition of HSP70 leads to the downregulation of HSP90 while inhibition of HSP90 results in upregulation of HSP70[160,171,174]. It has been shown however, that simultaneous inhibition of both HSP70 and HSP90 leads to greater MM cytotoxicity compared to inhibiting HSP90 alone[160,171,174]. To this end, there has been increasing interest in developing inhibitors against heat shock factor 1 (HSF), the “master regulator”of the heat shock response that controls the expression of both HSP90 and HSP70[158]. In preclinical studies,several novel HSF1 inhibitors (e.g., CCT251236, KRIBB11) were found to induce MM cell death that was associated with the induction of the UPR[175].
Autophagy modulators currently in clinical trials
Several clinical trials have highlighted the potential role of autophagy modulators in the treatment of MM.For the purpose of consistency while discussing these trials, overall response rate (ORR) is defined as a partial response (PR) or better, and clinical benefit rate (CBR) is defined as stable disease (SD) or better.
Hydroxychloroquine/Chloroquine
Hydroxychloroquine (HCQ)/Chloroquine (CQ) have been clinically studied for their potential role in inhibiting autophagy. Although the exact mechanism of HCQ/CQ has not been elucidated, it is thought to function by alkalinizing intracellular compartments which disrupts the autophagic proteolytic process[176].HCQ/CQ have been extensively studied in myeloma to potentiate the effects of other anti-myeloma drugs,particularly PI.
A phase I clinical trial assessed the efficacy of HCQ in combination with bortezomib in patients with relapsed or refractory MM[177]. Patients were given a 2-week run in with HCQ alone, followed by combination therapy with bortezomib. The combination of HCQ and bortezomib was well tolerated, with no adverse events meeting the criteria of a dose-limiting toxicity. Of the 22 patients assessed at the end of the study, 13.6% had a very good partial response (VGPR), 13.6% had minimal response (MR), and 45.5%had stable disease (SD). Interestingly, all of the patients who had a VGPR were bortezomib-naïve. Twentyseven percent of patients in the study had progressive disease (PD). Of these, two thirds were bortezomibrefractory. This study also found a therapy-associated increase in autophagic vesicles in BM plasma cells.However, due to the small sample size, the authors were unable to correlate number of autophagic vesicles with clinical response.
A smaller phase II trial looked at CQ in combination with bortezomib and cyclophosphamide in refractory MM[178]. Of the 11 patients enrolled, 8 completed at least 2 cycles and were assessed for clinical response. The most common side effects included fatigue, constipation, myalgia, anorexia, anemia and thrombocytopenia, but was generally well tolerated. 37.5% of patients achieved a PR with a median duration of response of 4 months. One patient (12.5%) had SD, and 50% of patients experienced PD, with an overall clinical benefit rate of 40%. These studies demonstrate that HCQ/CQ are well tolerated in combinations with PI and may help potentiate the effects of existing myeloma therapies.
A recent study looked at dual autophagic inhibition via HCQ and rapamycin with cyclophosphamide and dexamethasone in patients with relapsed or refractory MM based on their preclinical, synergistic antitumor activity[179,180]. The quadruple therapy was generally well-tolerated with one case of hematologic dose limiting toxicity (thrombocytopenia) occurring at a HCQ dose of 800 mg, which was then established as the maximum tolerated dose. Of the 18 patients enrolled in the trial, 1 had a VGPR and 3 had a PR, for an ORR of 22%. Seven patients had MR, and 5 had SD, for a CBR of 89%. Median duration of a response was 4.5 months, and median time to best response was 1.9 months. Two patients had immediate progression of disease. Unfortunately, correlative studies were unable to predict the depth of treatment response based on the number of autophagic vesicles in BM-derived MM cells. The promising results of this study warrant further investigation of dual autophagy modulation in MM.
HDAC6 inhibitors
HDAC inhibitors represent an important new group of anti-cancer drugs. There are 18 different isoforms of HDACs in human cells that are subdivided into 4 classes based on subcellular localization and noncell-based enzymatic activity[181]. Clinical studies with pan-HDAC inhibitors vorinostat and panobinostat outlined a narrow therapeutic window due to frequent and often severe hematologic and non-hematologic(particularly gastrointestinal/GI) toxicities, eliciting clinical interest in isoform-specific HDAC inhibition[182].
Ricolinostat is an oral, selective HDAC6 inhibitor that has been extensively studied in MM. A phase 1b multicenter trial of escalating doses of ricolinostat with lenalidomide and dexamethasone in patients with relapsed or refractory MM showed 55% ORR[182]. In lenalidomide-naïve and lenalidomide-sensitive patients,the ORR was 69%, compared to 25% in lenalidomide-refractory patients. In patients that responded, the median time to response was 7 weeks with a median duration of response of 24 months. Treatment with ricolinostat was overall well tolerated with 2 patients experiencing a dose-limiting toxicity at the highest tested dose which included syncope and myalgia.
Similarly promising results emerged from a phase 1/2 clinical trial assessing the safety and efficacy of ricolinostat with bortezomib and dexamethasone in patients with MM relapsed or refractory to PI,immunomodulatory drugs, or stem cell transplant[183]. In this study, 15 patients were recruited for dose escalation monotherapy with ricolinostat, and 57 patients were given combination therapy. Monotherapy with ricolinostat was well tolerated with no dose-limiting toxicities up to 360 mg Q.D. Patients on combination therapy did not have any dose-limiting toxicities during escalation studies. Most common side effects associated with combination therapy were gastrointestinal toxicities, cytopenia, and fatigue.Of the patients treated with monotherapy, 6 patients (6/15, 40%) had stable disease with a median response duration of response of 11 weeks. Patients on combination therapy had an ORR of 29%, and a clinical response rate of 39%. Interestingly, patients on combination therapy who were previously refractory to bortezomib had an ORR of 14%. The response seen in patients with bortezomib-refractory MM suggests that HDAC6 inhibitors can overcome resistance to PI.
Taken together, these results demonstrate a promising role for HDAC6 inhibitors in the treatment of relapsed and/or refractory MM. There are currently ongoing trials assessing the role of HDAC6 inhibitors in combination with anti-myeloma treatments such as pomalidomide and dexamethasone (NCT01997840,NCT02189343).
HSP90 inhibitors
A phase 2 clinical trial looked at the HSP90 inhibitor tanespimycin in combination with bortezomib in patients with relapsed and/or refractory MM[184]. All patients enrolled in the study experienced at least one side effect, with most common grade 3/4 toxicities being fatigue, thrombocytopenia, neutropenia, and abdominal pain. Four patients had significant liver toxicity with higher doses of tanespimycin, however this was manageable and reversible. The best responses observed to treatment were 1 MR in the 340 mg/m2group, and 2 PRs in the 175 mg/m2group. An additional 10 patients across all treatment groups had stable disease. A later phase 3 study evaluating tanespimycin and bortezomib has been completed, however results are not yet available.
KW-278 is a novel, non-anisomycin, non-purine based HSP90 inhibitor that has a more favorable pharmacokinetic and safety profile compared to tanespimycin[185]. A phase 1 trial examined the safety and efficacy of KW-278 in B-cell malignancies, including 22 MM patients[186]. The most common side effects included diarrhea, headache, rhinitis, and fatigue. Ten patients experienced grade 3 and 4 toxicities such as lethargy, syncope, QT prolongation, and neutropenia. Six patients experienced eye disorders (decreased visual acuity, blurry vision, dry eyes) that were deemed related to KW-278. All of the eye disorders were reversible with the exception of dry eyes. Two patients died during the trial and both deaths were determined to be unrelated to the study medication. Of the 21 MM patients evaluated, 20 patients (95%)had stable disease, and 1 patient had progressive disease.
A subsequent phase 1/2 study evaluated KW-278 with bortezomib in patients with relapsed or refractory MM who had failed at least 1 previous treatment[187]. The most common side effects were fatigue and gastrointestinal toxicity, while the most common grade 3-4 side toxicities were cytopenias. There was no sign of significant ophthalmologic side effects in the 95 patients enrolled in this study. There were 28 serious treatment-related adverse events such as lung infections, GI toxicity, anemia, hematuria, syndrome of inappropriate diuretic hormone, pancreatitis, and transient ischemic attack. Efficacy of the treatment was based on a KW-278 dose of 175 mg/m2with bortezomib at the standard concentration. The ORR was 39%, the CBR was 92%. Median progression-free survival was 6.8 months, and median duration of response was 5.6 months. Patients who were lenalidomide-naïve had an ORR of 45.5 as compared to 25%for patients previously exposed to lenalidomide. Bortezomib-naïve patients had an ORR of 44%vs. 33% in patients previously exposed to bortezomib. These findings suggest a potential therapeutic benefit of KW-278 with bortezomib in the treatment of relapsed or refractory MM.
PI3K-AKT-mTOR inhibitors
The PI3K-AKT-mTOR inhibitor everolimus was tested as a single agent in a phase 1 trial in patients with relapsed and/or refractory MM[188]. Most side effects were mild to moderate with gastrointestinal upset,elevated muscle and liver enzymes, and cytopenias being the most common. One patient developed an atypical pneumonia that was thought to be drug-related. Of the 15 patients assessed, 10 (67%) experienced clinical benefit with one patient achieving a partial remission after 4 cycles.
A phase I trial assessed the safety and efficacy of everolimus in combination with lenalidomide in patients with relapsed or refractory MM[189]. Most common side effects were fatigue, cytopenia, diarrhea and neuropathy. One patient discontinued treatment due to non-infectious pneumonitis that was related to everolimus, and another discontinued due to grade 3 myalgia. Of the 23 patients that completed 2 cycles of treatment, the CBR was 74% with 1 CR and 4 PR. In patients who were lenalidomide-naïve, the ORR was 90%, compared to 37.5% for patients who had previously received lenalidomide. Median progressionfree survival was 5.5 months, and median overall survival was 29.5 months. Promising results from these studies demonstrate the potential for mTOR inhibitors to be used the treatment of relapsed or refractory MM. A clinical trial looking at the role of mTOR inhibitors in combination with pomalidomide and dexamethasone is currently enrolling (NCT03657420).
Repurposing of FDA-approved drugs for use in MM
Metformin, the oral biguanide drug used in the treatment of diabetes, has been shown to exert anti-MM effects bothin vitroandin vivo. The exact mechanism of metformin’s anti-tumour activity has not been elucidated, but it is hypothesized to induce autophagy through the inhibition of STAT3 and BCL-2[132]. The anti-retroviral drug nelfinavir has also shown anti-MM effectsin vivoby triggering the UPR, and has been studied extensively clinical trials for relapsed MM[190,191]. A new phase 1 clinical trial is assessing the safety and efficacy of metformin, nelfinavir and bortezomib in with relapsed or refractory MM (NCT03829020).This is only one example of the potential utility of drug repurposing strategies in cancer, rationally designed based on theoretical synergistic mechanisms of activity.
CONCLUSION
Autophagy plays a crucial pro-survival role in MM. Increased protein synthesis and proteotoxic stress are hallmarks of cancer, and MM is the prototypic cancer with impaired protein homeostasis based on its staggering rate of immunoglobulin synthesis and baseline level of proteotoxicity. Importantly, autophagy protects MM cells from excessive ER stress by limiting the secretion of immunoglobulins and providing an alternative proteolytic pathway for the clearance of ubiquitinated proteins. Consistent with a therapeutic role in targeting protein homeostasis, PI are highly effective anti-MM agents that are FDA approved for the treatment of MM in all its stages. However, clinical resistance to PI is inevitable, leading to research interest in targeting alternative pathways contributing to protein quality control such as autophagy, alone or in combination with PI. As with other mechanisms of protein homeostasis, such as the UPR; while a basal level of autophagy is cytoprotective and inhibiting autophagy to blunt non- proteasomal proteolysis has also been shown to have therapeutic benefit, sustained autophagy as in the setting of persistent proteasome inhibition, may result in autophagic cell death. This highlights the “Janus-faced” role of autophagy in MM.Autophagy therefore represents an opportunity to therapeutically exploit MM’s unique “Achilles’ heel”: its dependence on protein quality control. With a better understanding of the molecular sequalae of autophagy inhibition/induction in MM, we are eagerly awaiting the development or repurposing of drugs to target autophagy in an effort to overcome PI resistance and improve outcome for our patients with MM.
DECLARATIONS
Authors’ contributions
All authors contributed to the writing of this review article.
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2019.
