
A new view of the mammary epithelial hierarchy and its implications for breast cancer initiation and metastasis
2019-07-29 09:32:50LindseyAnstineRuthKeri
Lindsey J. Anstine, Ruth Keri,2
1Department of Pharmacology, Case Western Reserve University, Cleveland, OH 44106, USA.
2Department of Genetics and Genome Sciences and Department of General Medical Sciences-Oncology, Case Western Reserve University, Cleveland, OH 44106, USA.
Abstract
Keywords: Mammary gland, mammary epithelial cells, breast cancer, stem cell
INTRODUCTION
Many of the processes that occur during mammary gland morphogenesis are appropriated during the formation and malignant progression of breast cancer. These include the regeneration and hormonally-driven proliferative expansion of the mammary epithelium, remodeling of the extracellular matrix and invasion into surrounding stroma, aversion of apoptosis, and recruitment of vasculature[1,2]. Thus, understanding the endocrine, cellular, and molecular mechanisms that control mammary gland development should provide critical insights into the processes that also underlie breast cancer growth and metastases and reveal novel approaches for preventing and treating aggressive disease. This review focuses on recent advances in our understanding of the development of the mammary epithelial cell hierarchy, revealing the abundant progression of cell states, many of which could undergo malignant transformation and give rise to the heterogeneous disease known as “breast cancer”.
The mammary gland is a uniquely dynamic organ that undergoes extensive remodeling throughout life in response to cytokine signaling and hormonal cues[3,4]. The primitive mammary epithelium is first detected around embryonic day 11.5 (E11.5) in the mouse. Between embryonic day 16 and embryonic day 18 (E18), this rudimentary structure invades the mammary fat pad, forming a primitive ductal structure. During puberty, the mammary epithelium expands to form a branched, ductal network that invades the stroma until it reaches the edge of the mammary fat pad. This ductal structure is maintained into adulthood where it remains highly flexible in response to oscillating levels of ovarian hormones during the estrous cycle, undergoing extensive proliferation and apoptosis[5]. In pregnancy, a unique hormonal milieu triggers the formation of secretory alveolar structures to form at the terminal end of each duct. Alveolar units function to produce milk proteins during lactation. Once lactation has ceased, the gland undergoes a process of involution in which massive architectural remodeling occurs, resulting in an epithelial structure that resembles the pre-pregnant state. Although morphologically similar, genetically engineered mouse models permitted tracing of “terminally” differentiated lactating cells and revealed that these cells contribute to the remodeled post-involuted gland and are molecularly distinct from their virgin counterparts[6,7]. The events of expansion, differentiation, and repopulation of the mammary tree reoccur with each subsequent pregnancy indicating extensive plasticity of the cells that comprise the gland. Closer examination reveals that the mammary gland consists of two main epithelial lineages, the inner luminal lineage and the outer myoepithelial lineage. The luminal lineage can be further subdivided into mature ductal and milk-producing alveolar cells. Myoepithelial cells are commonly referred to as “basal” since they are located on the basal side of the luminal cells[8]. These cells are highly contractile, and function to facilitate the movement of milk though the ducts.
The ability of the gland to undergo successive rounds of reconstruction in response to multiple pregnancies implicates the existence of a mammary stem cell (MaSC) population capable of generating all mature cell types within the gland. Studies utilizing transplantation and lineage tracing techniques further established that mammary epithelial differentiation is organized in a hierarchical, branched pathway wherein multipotent MaSCs give rise to a series of downstream intermediates[8-10]. With each differentiation step, cells become increasingly lineage-restricted moving from multi- to bi- to uni-potent progenitor states until terminally differentiating into mature epithelial populations.
Discoveries over the last 50 years have been integral to our understanding of the mammary epithelial cell hierarchy; however, the advent of single-cell (sc) sequencing technologies coupled with microfluidic cell sorting has recently uncovered intricacies in lineage relationships and heterogeneity of the mammary epithelium. Recent evidence has demonstrated that each cell is unique in its transcriptomic signature[11-14]. This revelation has redefined the differentiation cascade as a continuous trajectory of heterogeneous populations passing through gradual transcriptional states. These studies suggest that historical, rigid, models of the epithelial hierarchy should be replaced by a model that incorporates the diverse transcriptional profiles and paths represented throughout development, underscoring the immense diversity and flexibility that likely underlies mammary gland development and gives rise to breast cancer heterogeneity. Here we discuss the recent studies utilizing scRNA-sequencing of mammary epithelium and how these new findings have shifted our current perspectives of mammary gland development and breast cancer.
HISTORICAL PERSPECTIVE OF THE MAMMARY EPITHELIAL HIERARCHY
In 1959, DeOme and colleagues developed the mammary transplantation assay[15]. Using this technique, transplantation of murine ductal fragments[16,17]or dissociated suspensions of normal mammary epithelium[18]into the cleared fat pad of a recipient mouse resulted in the regeneration of a complete ductal structure. Smith and Medina then reported that morphologically distinct cells with regenerating capacity could be found throughout the entire mammary tree and persisted throughout pregnancy, lactation, and involution[19]. Both the frequency and regenerative ability of these cells was not affected by age or reproductive history[19,20]. Later studies demonstrated that mammary gland reconstitution was achievable upon transplantation of a single cell. This was first reported by Kordon and Smith, using serial transplantation and limiting dilution assays of mammary epithelium labeled with unique viral insertions generated by mouse mammary tumor virus (MMTV)[21]. Similarly, by labeling cells with a LacZ transgene, Shackletonet al.[22]confirmed this finding and demonstrated that stem cell populations were enriched in the basal epithelial fraction (defined by Lin-CD29hiCD24+cell surface markers). Additionally, cells from reconstituted glands harbored the same repopulating activity of the original and were capable of fully differentiating into milk-producing alveolar cells during pregnancy. These studies implicated the existence of a long lived multipotent mammary stem cell population (MaSC), capable of giving rise to all downstream lineages within the gland.
Following the disclosure of MaSCs, the search for molecular markers specific to the stem cell population became a major focus within the field. Transplantation of luminalvs. basal epithelial populations revealed that cells exhibiting stem-like properties were located within the basal fraction and could be enriched by FACS sorting using CD49fhiCD29hiCD24+/modEpCAM+Sca1lowexpression as markers[22-25]. Additionally, expression of s-SHIP[26], LGR5[27-29], Lrp5/6[30], and Axin2[31]enriched for populations with repopulating activity upon transplantation; however, stem cell activity was not exclusive to these subsets and the degree to which these markers may enrich for overlapping stem cell populations was not clear[10]. Difficulty surrounding the identification of MaSC markers was due, in part, to the rarity of MaSCs in the adult gland. The frequency of repopulating cells was found to be higher during embryogenesis (~7% of total cells by E18 compared to ~2%-5% frequency in the adult gland)[22,23,32,33]; however, enriched populations of fetal (f)MaSCs displayed altered gene expression profiles from their adult counterparts further complicating the isolation and characterization of a multipotent population[32].
Transplantation studies demonstrated the existence of a stem cell entity that was capable of multi-lineage differentiation within the adult gland. Yet, various studies employing lineage tracing analyses led to confusion surrounding these findings. While some groups provided evidence to support multipotent cells, others reported that only unipotent cells remain after birth. Early lineage tracing experiments by Van Keymeulenet al.[34], used the promoters of the keratin 14, 5, and 8 (K14, K5, K8) genes, to drive tracer expression and track both luminal (K8) and myoepithelial (K14, K5) lineages. This analysis demonstrated that prior to birth, K14+ cells were multipotent; however, after birth, these cells became unipotent, only giving rise to myoepithelial cells. Cells labeled by the K8 promoter revealed a second unipotent population responsible for maintaining the luminal lineage after birth. These unipotent populations supported epithelial expansion during puberty and pregnancy underscoring their long lived capacity within the gland. Additionally, lineage tracing at the single cell level revealed that unipotent populations were dispersed throughout the ductal tree, estimating that within each major duct, ~20 luminal and ~15 basal unipotent cells contribute to ductal expansion during puberty[35]. Furthermore, by employing clonal analyses at saturation using DOX-inducible systems to trace luminal (K8rtTA-CreTetO/Rosa-TdTomato) and myoepithelial (K14rtTA-CreTetO/Rosa-TdTomato) lineages, Wuidartet al.[36]also reported unipotent progenitors as solely responsible for maintenance of adult lineages. Evidence of luminal unipotent progenitors that are restricted to the alveolar fate has also been reported. In this case, the “parityinduced mammary epithelial cell” (PI-MEC) population consists of alveolar-like cells that originate during a female’s first pregnancy[6]. Unlike the majority of alveolar cells, PI-MECs do not undergo apoptosis during involution. Rather, these cells remain as a residual population within the parous gland and serve as alveolar precursors during subsequent pregnancies[6,37].
Contrary to these results, some genetic fate-mapping studies have supported the existence of multi- and bi-potent mammary epithelial cell populations within the adult. For example, tracing of the Wnt target protein C receptor (Procr) illustrated the presence of a multipotent cell within the basal layer of the adult gland that was capable of giving rise to all mature lineages[33]. Furthermore, studies have also reported bipotent progenitors in the adult gland. By combining a multi-color Cre reporter with three dimensional imaging techniques, Rios and colleagues traced luminal (ELF5+) and basal (Keratin 5+) cells during puberty and adulthood[28]. This analysis resulted in the tracing of discreet clonal patches of luminal or basal cells as well as patches containing both lineages, indicating a common cellular origin[28]. Similarly, an inducible Cre reporter mouse line, driven by the Axin2 promoter, labeled both bipotent and unipotent cells within the adult gland[38]. Further complicating these studies, Axin2+ cells can undergo cell fate switching[38]. This phenomenon has also been reported in mammary epithelial cells that are positive for Lgr5[29]. Interestingly, Axin2+ unipotent cells are basally-restricted during adulthood but become bipotent during pregnancy, suggesting that cell fate switching may occur in response to hormonal cues[10,38]. Pregnancy-induced bipotent mammary epithelial cells were also recently reported in a study by Songet al.[39]. Lineage tracing of K8+ luminal cells revealed bipotent luminal cells that gave rise to basal progeny during pregnancy or upon stimulation with estrogen or progesterone. Additionally, transplantation of luminal-derived basal cells into cleared fat pads of recipient mice, demonstrated their capacity to generate new mammary gland structures with normal morphology.
The conflicting results generated from transplantation studies with those from lineage tracing has been a source of controversy in the field. The primary question still remains: does a multipotent stem cell exist after birth as indicated by transplantation assays; or are progenitor populations solely responsible for postnatal development? Inherent flaws within transplantation and lineage tracing methods likely contribute to the conflicting evidence within the field.
Transplantation assays have been highly scrutinized since this method includes the dissociation of mammary epithelial cells from their normal tissue architecture followed by their reintroduction into a new mammary tissue environment that has undergone invasive surgical resection. The drastic micro-environmental changes that epithelial cells must endure for this procedure has raised concerns that this assay may impart multipotent potential onto cells that would otherwise be restricted to specific differentiation outcomes[10]. For example, Wnt/-catenin-responsive cells only give rise to myoepithelial cells during puberty and pregnancy, however; upon transplantation these cells can regenerate both luminal and myoepithelial lineages[38]. Additionally, in lineage tracing experiments, K14+ cells are restricted to the myoepithelial lineage after birth; however, transplantation of K14-labeled cells from 4-week old mice results in the formation of new ductal structures containing both myoepithelial and luminal cells[34]. The ability of transplantation to alter stem cell fate has also been demonstrated in other systems, including hair follicle development[40]and hematopoiesis[41]. It is plausible that differences in the microenvironment such as stromal, hormonal, and inflammatory cues, may impart phenotypic changes in mammary epithelial cell populations, activating primitive precursor pathways in their lineage. This may be especially relevant in the case of mammary transplants as the surgical resection needed to clear the innate epithelia likely elicits a “wound healing” response in the local environment of the transplant. Thus, transplantation assays may not accurately represent stem cell biology as it would intrinsically occurin vivo.
Although lineage tracing allows for the visualization of cellular differentiation within the context of the normal tissue environment, it is not without its own set of limitations. The ability to detect rare heterogeneous populations of cells using genetic lineage tracing is highly dependent on recombination efficiency. High variability in the extent of recombination not only exists between mouse models (i.e., K5-Cre versus K14-Cre) but also between individual mice from the same strain[42]. In addition, labeling efficiency varies significantly throughout the ductal tree of a single mammary gland[42]. Several explanations for this variability have been postulated[8,10,42]. Inadequate strength and specificity of the promoter driving recombination could result in inconsistent labeling. This may be particularly relevant to quiescent stem cells that are transcriptionally uncommitted to a certain lineage. Additionally, inducible creERT2 models are reliant on accurate dosing of Tamoxifen, a selective estrogen receptor (ER) modulator, to induce sufficient recombination. Since mammary gland development is highly dependent on ER signaling, tamoxifen administration, even at low does, can alter normal mammary gland biology and lead to false readouts[43].
Taken together, the results from both transplantation and lineage tracing studies have led to a highly compartmentalized view of the mammary epithelial hierarchy in which fetal MaSCs give rise to a common bipotent progenitor which differentiates into lineage-restricted and unipotent luminal and myoepithelial progenitors. Unipotent progenitors then give rise to mature myoepithelial and luminal lineages (for detailed reviews see Visvader and Stingl[10], 2014, and Yanget al.[44], 2017) [Figure 1]. Although transplantation and lineage tracing methods have been fundamental to our understanding of mammary epithelial biology, it has become increasingly evident that these methods are insufficient to fully characterize rare or highly heterogeneous cell populations. Gene expression signatures on bulk isolates of the major epithelial compartments including luminal progenitors, mature luminal, and MaSC-enriched basal epithelial cells, have been generated[45]. However, the utility of these signatures to identify critical molecular regulators and markers of epithelial subsets has been limited due to the heterogeneity of each epithelial population in which only a small fraction of cells expresses the same set of transcripts (as in the case of stem populations within the basal fraction)[22,32,46]. As a single cell can give rise to an entire mammary gland, it is imperative that the molecular states and instructive signals occurring at the single cell level be examined to fully grasp how individual cells traverse the differentiation cascade as well as discern how specific cell states can re-emerge in breast cancer and contribute to metastasis.
A NEW VIEW OF THE MAMMARY HIERARCHY
Single-cell sequencing has emerged as a powerful tool capable of uncovering the transcriptional heterogeneity of diverse cell types within a tissue[47]. Although transcriptome profiling of single cells was reported over 20 years ago[48,49], only recently has it become a widely utilized technique. Technological advances in areas such as microfluidics and in situ barcoding have allowed for high throughput, cost effective sequencing of hundreds to thousands of cells within a given sample[50]. Furthermore, the ability to analyze large numbers of cells at high resolution has mandated the development of innovative approaches for downstream data analysis. Software tools have recently become available to infer evolutionary history and lineage relationships within heterogeneous cell populations[51].
Recently, several independent laboratories have utilized scRNA-seq to identify epithelial subpopulations and their differentiation trajectory within the mammary gland[11-14]. Together these studies have examined the complete mammary developmental cycle encompassing embryonic through adult stages and pregnancy through post-weaning. The transcriptional landscape generated from these studies has provided critical insight into epithelial differentiation, transforming the way in which we view the mammary epithelial hierarchy. The new model emerging from these studies emphasizes the fluid and gradual progression of epithelial differentiation and reveals a greater degree of cellular heterogeneity than had previously been acknowledged.
SINGLE-CELL SEQUENCING OF THE MAMMARY EPITHELIUM
Prior to birth, sc-RNA sequencing has shown that the epithelium consists of a continuum of primitive cellular states in which unique subpopulations could not be detected[12]. This supports previous findings generated from gene expression analysis of bulk fMaSCs, demonstrating that fMaSCs are comprised of a single CD24+CD49f+ population[32]. During embryogenesis, the majority of mammary epithelial cells exhibit dual expression of both luminal and myoepithelial signature genes[12,32,52]. This hybrid gene expression signature is associated with multipotent potential and a loss of dual lineage expression as cells become restricted to unipotent potential[52].
At birth, many mammary epithelial cells maintain a hybrid gene expression profile[12]. This dual lineage signature may include rare bipotent progenitor populations previously observed by lineage tracing studies[28,38]. However, the majority of this population likely represents cells spanning various transitional stages of lineage restriction. Cells become increasingly more lineage restricted throughout puberty; however, the precise timing of the major bifurcation event between luminal and basal cells remains unclear. In work by Palet al.[53], scRNA-sequencing of mammary epithelium from 2 (pre-puberty), 5 (puberty), and 10 (adult) week-old mice demonstrated that before puberty, the mammary epithelial cell populations are largely homogeneous, expressing a basal (myoepithelial)-like gene signature. Upon puberty, a fraction of cells upregulate expression of luminal genes such as Epcam, and Keratins 8, 18, and 19, suggesting that bifurcation initiates at this time[53]. However, Giraddiet al.[12], specifically assessed changes in the early postnatal epithelium by examining glands from mice at day zero (PN0) and day 4 (PN4) after birth. In contrast to the report by Pal and colleagues, they found that two distinct cellular populations, one containing myoepithelial cells and the other containing luminal precursors, begin to emerge as early as PN4. While these studies confirm that the mammary epithelium remains in a primitive state until postnatal stages during which lineage restriction commences, the specific timing of those commitments remains unclear.Sc-RNA sequencing of adult stages clearly demonstrate that mature epithelial cells are transcriptionally segregated into basal (myoepithelial) and luminal linages[11,12,53]. Within these overarching phenotypes, up to 15 transcriptionally distinct clusters (11 luminal and 4 basal) have been reported across nulliparous and reproductive stages[11]. However, the majority of these subpopulations cannot be characterized by expression of a single gene, indicating that cells remain highly heterogeneous and many of these clusters may represent transient states of differentiation. The luminal lineage within the nulliparous gland undergoes multiple differentiation events. Luminal progenitors give rise to a series of intermediate states that bifurcate to generate a hormone responsive lineage (ER+) and a secretory alveolar lineage (ER-)[11,12,14]. Upon pregnancy, ER- cells undergo massive expansion to produce the alveolar cells needed for lactation. The expanded population expresses genes associated with proliferation, fatty-acid transport, and lipid biosynthetic and homeostatic processes[11]. Recent lineage tracing studies have proposed that ER+ and ER- luminal lineages arise from independent progenitor populations[54,55]. In work by Wanget al.[54], PROM1+ progenitors were found to give rise to ER+ luminal cells whereas SOX9+ progenitors give rise to ER- cells. Thus, as heterogeneous luminal populations undergo gradual differentiation, the acquisition of SOX9 or PROM1 expression may be a major determinant of further luminal cell fate specification.
STEM AND PROGENITOR POPULATIONS DETECTED BY SINGLE-CELL RNA SEQUENCING
The sc-RNA-sequencing studies reported to date have not detected a single transcriptionally distinct multipotent stem cell population within the adult murine gland[11,12,53]. However, dormant populations that possess stem potential under specific environmental cues may exist. In a study by Sunet al.[13]mammary epithelial stem cell markers including Bcl11b[56], Tspan8[57], and Procr[33]were detected in the adult gland; however, expression on these genes was mutually exclusive, indicating each labels an independent population. Further analysis of a novel population marked by Cdh5+, revealed increased stem activity within this group of cells as demonstrated by both mammosphere andin vivotransplantation assays[13].
In human breast, bipotent progenitor populations within the adult gland have been identified in cells isolated from reduction mammoplasties[14]. Using Monocle software analysis of sc-RNA sequencing data collected from seven individuals, Nguyenet al.[14], mapped both luminal and myoepithelial lineages to a common cellular origin. These cells exhibited a basal gene expression signature as well as expression of ZEB1. This contrasts with the inability to identify subpopulations of bipotent progenitors using sc-RNA sequencing of mouse mammary glands. Discrepancies between mouse and human studies may be due to differences in the methodologies employed to generate scRNA-seq profiles [Table 1]. These include epithelial cell isolation methods, read depth, and data analysis and interpretation. For example, in work by Giraddiet al.[12](2018) and Bachet al.[11](2017), sc-RNA sequencing clustering results were based on epithelial cells that were positive for EPCAM expression. Thus, there remains the possibility that an EPCAM-stem cell population was overlooked in the murine studies. Additionally, cells double positive for the major luminal and basal cell markers, cytokeratin 8 and 14 respectively, are detected within the basal epithelial layer of human adult breast tissue[14]. This is in contrast with mouse basal epithelium where double positive cells are not detected after birth[22]. This difference in dual cytokeratin expression coupled with the interpretation that cells expressing both luminal and basal markers represent bipotent progenitors may contribute to the differences between mouse and human scRNAseq findings. Furthermore, evidence from transplantation assays suggests that stem populations are widely distributed within the gland[16,17,19]; however, it is unknown if specific subpopulations are spatially restricted. In this regard, it has been reported that a group of highly quiescent MaSCs, marked by Tspan8 expression, are located within proximal regions of the adult murine gland, closest to the nipple[57]. Thus, discrepancies between data sets may also arise depending on area of the mammary gland from which the sequenced cells were collected.
Luminal progenitors that are first identified as mixed lineage cells gradually gain luminal orientation during postnatal development[53]. By puberty, a distinct luminal progenitor population can be detected. However, scRNA-sequencing has further uncovered novel subcategories within the greater luminal progenitor population. For example, prior to puberty, CD55 is a marker of rare basal cells that give rise to a subset of luminal progenitors during adulthood[53]. CD55+ cells within the luminal fraction exhibit increased colony forming ability compared to CD55- cells[53]. In addition, populations positive for both CD55 and the progenitor marker, CD14 possess an even greater colony-forming ability. Luminal progenitors acquire additional complexity during pregnancy. Single-cell analysis of luminal progenitors in the nulliparous adult compared to post-involution glands revealed permanent transcriptomic alterations within a subset of postinvolution cells[11]. Following involution, luminal progenitors retain expression of genes associated with lactation and milk production. It is likely that this population overlaps with the previously described PIMEC population[6].
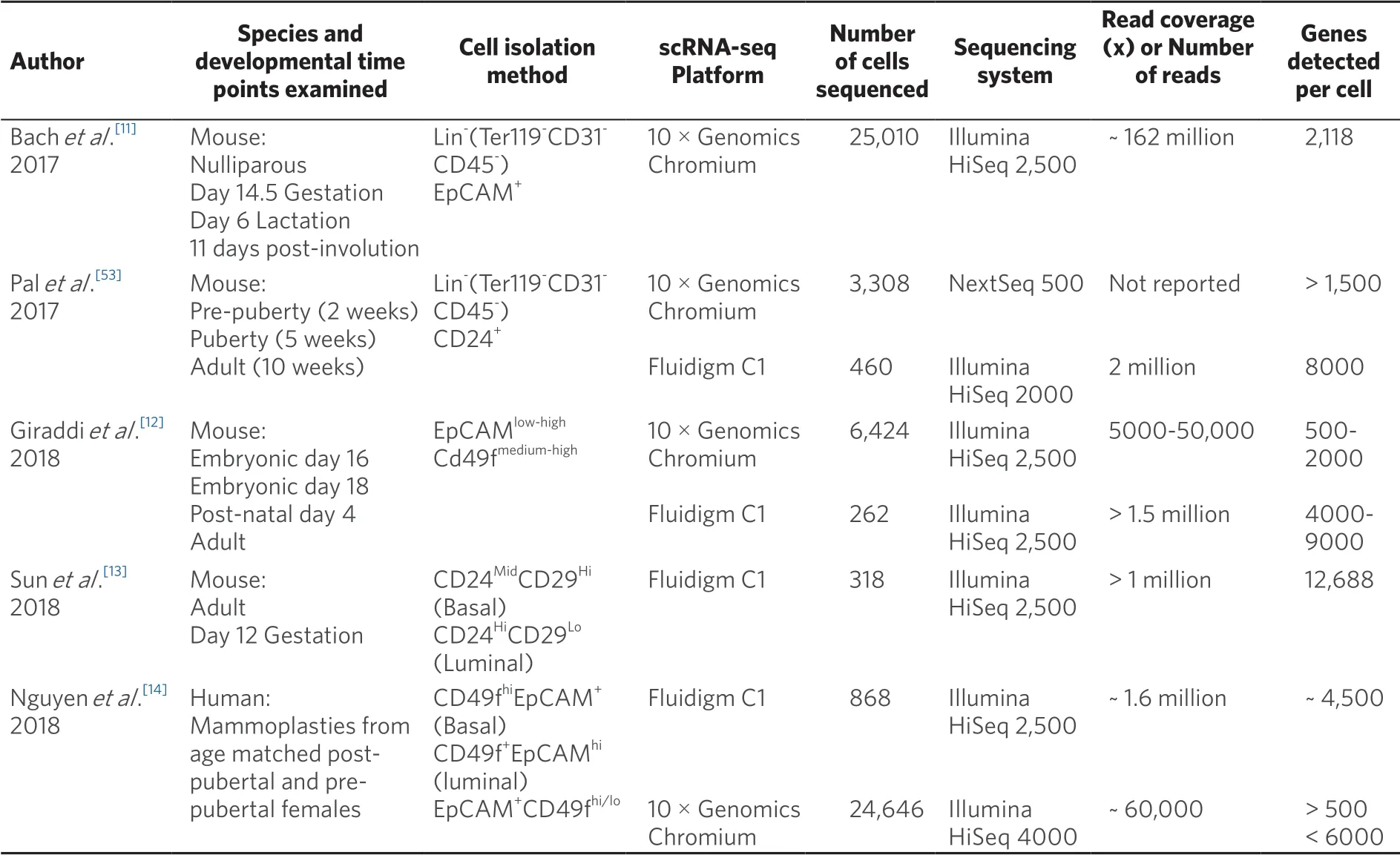
Table 1. Comparison of scRNA-seq methodologies used to examine the mammary epithelial hierarchy
CELLULAR PLASTICITY OF THE MAMMARY EPITHELIUM
The ability of mammary epithelial cells to express both luminal and myoepithelial signature genes at various stages of maturation and reproduction suggests that a fraction of cells may maintain lineage plasticity throughout life[12,14,52,53]. Studies investigating chromatin accessibility within the mammary epithelium at fetal and adult stages have demonstrated that bi-lineage gene expression is achievable due to epigenetic regulation of genes associated with multiple lineages[12,58]. In a recent study by Draviset al.[58], gene expression profiles and open chromatin regions were compared between fetal (f)MECs, luminal progenitors, and mature lineages using RNA-seq and ATAC-seq, respectively. fMaSCs expressed both canonical luminal and basal genes and gene loci were associated with open chromatin, reflecting their immature, multilineage state (consistent with Giraddiet al.[12]). While, the chromatin architecture of adult myoepithelial cells resembled that of fMaSCs in which luminal and myoepithelial gene loci were associated with open chromatin; only genes associated with the myoepithelial lineage were expressed. These results indicate that although myoepithelial cells express lineage-restricted genes, they retain the flexibility to adopt a luminal identity similar to multipotent fMaSCs[58]. This may explain the multipotent capacity of basal cells observed upon transplantation[22,23]. Several epigenetic factors have been shown to play a role in defining mammary epithelial cell fate. These include the histone methylation reader PYGO2[59], the polycomb-repressive complex-1 member, BMI1[60,61], and the polycomb-repressive complex 3 member, EZH2[62]. BMI1 and EZH2 also play key roles in metastasis of breast cancers, likely due to their activation of more primitive cell fates[63-66]. Yet how these players collaborate to orchestrate the epigenetic continuum that drives fate determination and heterogeneity is not fully understood.
Differential chromatin accessibility of both myoepithelial and luminal genes in basal cells may explain the ability of transcriptional drivers to induce cell fate switching in committed cells. In a recent example, overexpression of Notch1 in Smooth muscle actin (SMA)+ or K5+ myoepithelial cells, was sufficient to commit cells to a luminal fate[67]. Complementary to this, overexpression of p63 in luminal cells was sufficient to reprogram them into a myoepithelial state[52]. Although the mechanism of p63-mediated cell fate switching is unknown, chromatin remodeling is likely required as mature luminal cells exhibit repressed chromatin at basal gene loci[58]. SOX10 has also emerged as a major transcriptional regulator of epithelial cell fate[58]. Within fMaSCs, SOX10 motifs are enriched at accessible chromatin regions flanking highly expressed genes. Furthermore, tumors expressing high SOX10 levels exhibit neural crest-like features[58]and high SOX10 expression is correlated with the aggressive, basal-like breast cancer subtype. As embryonic neural crest cells are multipotent and highly mobile[68], SOX10 reprograming may lead to primitive cellular states that could contribute to aggressive tumor phenotypes.
A NEW MODEL FOR THE MAMMARY EPITHELIAL HIERARCHY
Cumulative evidence from the recent papers utilizing sc-RNA-sequencing has revealed that the historical models by which the mammary epithelial hierarchy has been traditionally represented do not accurately portray the complexity of the system[11-14,53]. Previous models in which cellular states are depicted as discrete populations that differentiate along restricted paths is an oversimplification [Figure 1]. Instead, recent results indicate that heterogeneous cell populations gradually advance, and likely also retreat, through a differentiation trajectory[11,12,53]. This is supported by the work from several groups, reporting a vast array of epithelial transcriptional profiles throughout the stages of mammary gland development[11-14,53]. Similar findings have redefined the hierarchical visualization of hematopoiesis[69]. Using the new visual representation of hematopoietic differentiation suggested by Laurenti and Gottgens as an example, we propose a similar paradigm to represent the mammary epithelial hierarchy [Figure 2]. As depicted in this model, groups of heterogeneous epithelial cells traverse through the differentiation landscape, passing through cellular states that have been previously defined including fMaSCs, luminal and myoepithelial progenitors, and mature luminal and myoepithelial lineages. The newly reported heterogeneity of these populations implies that each cell may take a slightly different transcriptional path from the next as it undergoes differentiation. Additionally, the array of cellular states detected within the gland at any one time suggests that variation in temporal dynamics of differentiation may exist between individual cells. Moreover, this model implies significant forward and reverse plasticity of cell states that could be impacted by the microenvironment. This could partially explain historical difficulty in attempting to isolate and characterize specific subpopulations; however, further studies are needed to test these predictions.
IMPLICATIONS FOR TUMOR INITIATION AND METASTASIS
Breast cancer is an amalgam of diseases that exhibit both inter- and intra-tumoral heterogeneity[70]. Gene expression profiling has led to the classification of five overarching subtypes, including luminal A, luminal B, HER2+, basal-like, and claudin-low[71-74]. However, within each subtype, tumors can exhibit further variability in gene expression, molecular function, and drug susceptibility conveying distinct patient outcomes. The heterogenic nature of breast cancer is thought to arise from the combination of cellular origin, genetic and epigenetic changes, and environmental context[1,10,70,75].
Molecular profiling studies have revealed that the gene expression patterns of cancer subtypes align with those of normal mammary epithelial cell lineages[76,77]. This suggests that tumor subtypes may originate from distinct mammary epithelial subpopulations. It is widely speculated that epithelial stem/progenitor populations may serve as tumor initiating cells since their longevity and ability to self-renew affords the accumulation of genetic mutations. This has been supported by gene expression profiling studies. For example, the gene expression profile of claudin-low tumors most closely resembles that of MaSC/basal epithelial cells, whereas basal-like tumors more closely resemble luminal progenitors[45,72,76,78]. Further evidence supporting luminal progenitors as the cell of origin for the basal-like subtype has been shown in tumors harboring BRCA-1 mutations[76,77,79]. More recently, sc-RNA-sequencing of human breast epithelial cells demonstrated that cells expressing luminal progenitor genes correlated most highly with basal-like breast cancers from the METABRIC dataset[14]. Despite these findings, not all cancer subtypes have been correlated with a cell of origin and the inability to isolate pure populations of mammary stem/progenitor cells has impeded functional testing. Moreover, it is also feasible that cancers acquire the transcriptional profiles of specific cell states due to the mutations that drive tumor development or progression and that the cells of origin may not be consistent with final tumor deposition. In this case, identifying the contextual cues that promote normal epithelial hierarchical progression would likely reveal pathways that are also activated or suppressed in tumors that promote their phenotypes.
The new view of the mammary epithelial hierarchy suggests that the continued search for a tumor cell of origin based on molecular profiles that have historically been used to characterize the major epithelial populations (mature luminal, myoepithelial, luminal progenitor ext.) is unlikely to reveal discreet cells from which different breast cancer subtypes emanate. The complexity of the normal epithelium suggests a similar heterogeneity is present in the primary and secondary tumors that arise from it. Indeed, a study comparing sc-RNA-seq profiles generated from triple-negative patient tumors revealed that a subset of cells within each tumor expressed profiles resembling a spectrum of epithelial differentiation statuses[80]. It now can be presumed that cells residing anywhere along the continuous differentiation trajectory may acquire tumorinitiating mutations [Figure 2] and that the location of the cell within the differentiation hierarchy will determine its transcriptional status and influence whether acquired mutations will be oncogenic. This is most consistent with the inability to identify a single lineage that contributes to HER2/Neu-induced tumors because amplification or overexpression of the ERBB2 gene may occur in an array of cell states rather than a single cell of origin for all tumors[81,82]. Similar findings have been reported in leukemia, in which progenitor cells rely on the activation of the β-catenin pathway for oncogenic transformation[83]. Although it is noted that some populations may be more susceptible to mutagenic events than others[84,85], the possibility that phenotypically similar tumors arise from cells residing in the exact same cellular state is slim. This notion is supported by the transcriptional heterogeneity observed across breast cancer patient tumors within the same subtype[86]. Several genetic mutations have been associated with the stratification of breast cancer subtypes. These include mutations in GATA3, RUNX1, or NCOR1 which are associated with the Luminal A subtype; whereas mutations in the tumor suppressor retinoblastoma, RB1, or CDH1 have been associated with luminal B tumors[10,87,88]. Although these mutations are associated with the acquisition of a specific breast cancer subtype, it is likely the combination of the preexisting epigenetic and transcriptional status of the cell along with the transforming mutations that explain tumor heterogeneity and give rise to the variable therapeutic responses observed between patients.
Accumulating evidence suggests that a rare population of stem-like progenitor cells are responsible for tumor initiation, progression, metastasis, and therapeutic resistance[89]. These cells are referred to as cancer stem cells (CSCs) or “tumor initiating cells” and share many characteristics with normal mammary epithelial stem cells including the ability to self-renew and differentiate. Additionally, CSCs share gene expression profiles that closely resemble those of primitive mammary epithelial cells, including the expression of stem and EMT associated genes[90]. This has led to many to posit that CSCs arise from mammary stem/progenitor cells, however definitive evidence for the origin of CSCs in breast cancer is lacking[91]. The extensive heterogeneity within the mammary epithelium that has been revealed by scRNA-seq makes it reasonable to presume that many cellular states have the potential to give rise to CSCs. Furthermore, the unique transcriptional and epigenetic context of an epithelial cell prior to transformation likely determines the aggressive nature of the arising tumor, including its ability to metastasize. Indeed, many studies have identified transcriptional regulators of normal mammary development that also encourage aggressive behaviors during cancer. This is the case for transcriptional drivers of stem cell and EMT programs including SOX9[92], SLUG[92], and SOX10[58]. In a study by Guoet al.[92], expression of both SOX9 and SLUG was found to be necessary for maintenance of the MaSC phenotype. Moreover, coexpression of SOX9 and SLUG in breast cancer cell lines was sufficient to induce stem cell-like properties, including activation of an EMT program and metastasis-seeding abilities. Similar results were observed for SOX10[58]. Binding of SOX10 within regions of open chromatin that flank genes associated with EMT or with embryonic neural crest cells (NCC) was found to promote migratory and invasive behavior within mammary tumor cells. Thus, it is possible that epithelial cells expressing high levels of transcriptional drivers of EMT and stem-like states at the time of transformation may be predisposed to malignant phenotypes. Likewise, the chromatin accessibility of genes associated with processes such as EMT and NCC may ‘prime’ cells for aggressive behaviors associated with CSC phenotypes.
The formation of CSCs and their ability to seed new tumors at distant sites is also highly dependent on the surrounding microenvironment[93-95]. Notably, contextual stimuli such as exposure to TGFβ, can initiate stem cell and EMT programs in normal mammary epithelial cells that then promote their transition to a CSC state[94]. The resulting CSCs remain highly susceptible to surrounding cues. This is especially relevant in the context of metastasis, in which Paget’s “seed and soil” hypothesis proposes that a tumor-permissive environment is required for disseminating tumor cells to engraft and metastasize into new tissue sites[93,96]. As breast cancer arises from the normal epithelium, the contextual cues regulating normal epithelial behavior are likely mirrored in breast cancer cells. Therefore, understanding the reciprocal interactions between epithelial cells and their niche should also inform the mechanisms underlying tumor initiation and metastasis.
In this regard, the recent studies using sc-RNA-sequencing outside the context of disruptive transplantation assays, have revealed that epithelial populations may be more highly influenced by the surrounding environmental cues than previously appreciated. As discussed earlier, previous studies using transplantation assays demonstrated the multipotent potential of rare mammary epithelial cells within the adult gland[22,23]. However, lineage tracing analyses were unable to replicate these results and recent sc-RNA-sequencing has further supported the lack of multipotent cells during adulthood. Thus, it has become increasingly apparent that introduction of isolated cells into wounded stromal environment may elicit cellular phenotypes that are not supported under normal physiological conditions. A similar phenomenon is likely to occur with breast cancer initiation and progression. While acquisition of an “oncogenic” mutation may occur in varied cells along the differentiation hierarchy, its impact on tumorigenesis and progression may rely upon the stromal microenvironment, particularly an environment that is reflective of wound repair such as that which occurs with high breast density[97], obesity[98], and in post-lactational remodeling[99]. The same is also likely to be true with metastasizing cancer cells. Instructional interactions between the epithelium and surrounding stroma are well established. This was most exquisitely demonstrated in extensive studies by Smith and colleagues revealing that the local microenvironment of the mammary gland is sufficient to reprogram progenitor cells from completely different tissues (including testicular and embryonic stem cells) into mammary progenitors that were capable of forming normal mammary gland structures[100-104]. Furthermore, alterations in the microenvironment may influence cellular transformation. In work by Barcellos-Hoff and Ravani[105], irradiating the cleared fat pads of recipient mice, lead to alterations in growth factor activity and extracellular matrix composition. Following irradiation, implantation of non-tumorigenic mammary epithelial cells resulted in tumor formation more readily in irradiated mice. Conversely, epithelial tumor cells injected into non-irradiated mammary fat pads reverted to form normal mammary gland ducts rather than tumors[106].
The effects of contextual cues on the epigenetic and transcriptional status of major transcriptional regulators has also been recently highlighted. Although SOX10 is highly expressed in cells isolated from MMTV-PyMT tumors, this expression was lost when cells were grown in 2D culture conditions[58]. The loss of SOX10 expression was associated with a switch from open to closed chromatin at the SOX10 locus, emphasizing the role of environmental cues on epigenetic status. This is not a new concept, as changes in the epigenetic landscape are known to occur in response to hormonal exposure[62,107].
CONCLUSION
The scRNA-sequencing studies reviewed here have provided a detailed transcriptional map of mammary epithelial differentiation. However, these data indicate that previously underappreciated complexity undoubtedly underlies the regulation of the heterogeneous transcriptional networks observed. The unique transcriptional status within each epithelial cell is likely correlated with a unique epigenetic profile and it is anticipated that these profiles have a considerable level of plasticity that is dictated by the microenvironment and, in the case of tumors, mutational status. Studies examining the chromatin accessibility within bulk isolations of epithelial populations (MaSCs, basal, luminal and luminal progenitors) have demonstrated that each population is associated with distinctive active and repressed regions, many of which are associated with functional consequences and specific tumor subtypes[12,58,107]. The emerging ability to simultaneously assess both the transcriptional and epigenetic status at a single cell level should allow for clearer understanding of the lineage relationships in the normal gland and in tumors as well as reveal whether tumor cells can readily transition through states as has been observed in the normal breast. It is important to note, however, that while single cell sequencing technologies can provide high resolution maps of cellular states, they cannot make definitive predictive connections to the resulting cellular fates. The use of newly developed technologies, such as “CellTagging” that allow for early transcriptional statuses to be linked to cellular fates will allow for a clearer understanding of epithelial lineage relationships within the mammary gland[108]. Furthermore, studies layering single cell technologies that assess transcriptional, epigenetic, and proteomic data within the normal gland, primary tumor, and associated metastatic lesions should provide important information regarding the extent of plasticity of primary and metastatic tumor cells that can be therapeutically leveraged to promote differentiation of breast cancers into less malignant states.
DECLARATIONS
Authors’ contributions
Conceptualized and drafted the manuscript: Anstine LJ
Revised the manuscript for important intellectual content: Keri R
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Both authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2019.
 Journal of Cancer Metastasis and Treatment2019年6期
Journal of Cancer Metastasis and Treatment2019年6期
- Journal of Cancer Metastasis and Treatment的其它文章
- The role of hypoxia-induced factor 1α in breast cancer
- Circulating microRNAs and liquid biopsy: murine xenograft models for technical validation of clinical protocols
- Paxillin serine 178 phosphorylation in control of cell migration and metastasis formation through regulation of EGFR expression in breast cancer
- Significance of trace eIement quantities in the prostatic secretion of patients with benign prostatic hyperpIasia and prostate cancer
- Cellular plasticity and metastasis in breast cancer: a pre- and post-malignant problem
- Differential expression and function of the endogenous lactate receptor, GPR81, in ERαpositive/HER2-positive epithelial vs. post-EMT triple-negative mesenchymal breast cancer cells