
QuEChERS EMR-Lipid技术结合LC/MS/MS快速筛查与确证猪肉中55种兽药残留
2019-07-10 13:20黄泽玮闵宇航刘忠莹
食品工业科技 2019年11期
黄泽玮,闵宇航,杜 钢,黄 瑛,王 颖,刘忠莹
(四川省食品药品检验检测院,四川成都 611731)
食品安全是食品工业健康快速发展的前提,而目前我国食品安全问题却不容乐观,其中兽药残留问题严重危害着公共卫生安全及人体健康[1]。兽药的种类有很多,主要包括促生长素、抗生素类、抗寄生虫类等多种药物,其在预防、治疗、诊断动物疾病中发挥重要作用。过量残留的兽药可通过食物链进入人体内,对人体健康造成损伤[2]。因此严格监控动物源性食品中兽药残留具有重要意义。
随着兽药滥用越来越严重,检验人员也面临巨大的挑战:每检验几个或一类化合物就需要更换一种前处理方法和检测方法,耗费极大的人力、物力、财力和时间。同时动物源性食品种类多,基质复杂且差异大,检测对象多,是食品中兽药残留处理和检测的难题。近年来,兽残检测的相关文献中,前处理方法主要有:固-液萃取法[3-4]、固相萃取法[5-7]、、基质固相分散技术[8-10]和QuEChERS[11-13]等,检测技术有:高效液相色谱法[14-15]、液相色谱-串联质谱法[16-17]、液相色谱-四极杆-飞行时间质谱法[18-19]等。其中近两年新推出的增强型除脂分散净化剂(Enhanced Matrix Removal-Lipid,EMR-Lipid)是一种根据油脂类成分及大多数目标分析物结构特点设计的增强型脂质去除的QuEChERS净化材料,对长链结构物质(如磷脂、游离脂肪酸、甘油三酯等)具有强大的吸附作用,能有效降低高脂肪含量基质对目标成分的干扰[20]。目前该技术还未见用于猪肉中多兽残检测的报道。
本文选择了55种易出现滥用的兽药(包括喹诺酮类抗生素、磺胺类抗生素、头孢类抗生素、β-受体激动剂、抗寄生虫药等)作为研究对象,以猪肉作为研究基质,运用QuEChERS EMR-Lipid结合液质联用,建立猪肉中55种兽药残留的快速检测方法。
1 材料与方法
1.1 材料与仪器
阿苯达唑-2-氨基砜、2-氨基氟苯咪唑、5-羟基甲苯咪唑、阿维菌素、阿苯达唑、阿苯达唑亚砜、阿苯达唑砜、金刚烷胺、噻苯咪唑酯、头孢孟多锂、头孢他美酯、头孢哌酮、头孢噻肟、头孢噻呋、环丙沙星、克伦特罗、氯羟吡啶、氯丙那林、丹诺沙星、双氟沙星、恩诺沙星、依普菌素、芬苯达唑、倍硫磷亚砜、氟罗沙星、氟甲喹、甲苯咪唑、氨基甲苯咪唑、萘啶酸、奥比沙星、奥芬达唑砜、奥苯达唑、恶喹酸、培氟沙星、喷布特罗、吡罗昔康、普萘洛尔、沙拉沙星、司帕沙星、磺胺苯酰、磺胺氯吡嗪、磺胺二甲氧嗪、磺胺二甲异噁唑、磺胺甲基嘧啶、磺胺二甲嘧啶、磺胺甲噻二唑、磺胺甲基异噁唑、磺胺甲氧哒嗪、磺胺间甲氧嘧啶、磺胺苯吡唑、磺胺喹恶啉、替诺昔康、三氯苯达唑、妥布特罗、泰乐菌素 标准品,阿尔塔科技有限公司,浓度为100 μg/mL,溶剂为甲醇;乙腈、醋酸铵 色谱级,飞世尔化学(Fisher Chemical);甲酸 优级纯,上海安谱科学仪器有限公司(Anpel);增强型除脂分散净化管(EMR-Lipid dSPE,1 g/15 mL)、增强型除脂萃取盐包(EMR polish,3.5 g)、陶瓷均质子 美国安捷伦科技公司(Agilent)。
1290-6460超高压液相色谱-三重四极杆质谱联用仪 美国安捷伦科技公司(Agilent);CP225D电子分析天平 德国赛多利斯公司(Sartorius);MULTIFUGE X3R高速冷冻离心机 美国赛默飞世尔公司(Thermo Fisher);TurboVap LV全自动氮吹浓缩仪 美国拜泰齐公司(Biotage);MS3漩涡混匀仪 德国艾卡仪器设备有限公司(IKA)。
1.2 实验方法
1.2.1 液相条件 色谱柱:Agilent Eclipse Plus C18(150 mm×3.0 mm,1.8 μm),流速0.5 mL/min。流动相为0.2%甲酸水溶液(A相)-0.2%甲酸乙腈(B相),梯度洗脱:0 min(A相98%:B相2%,后同)→0.5 min(98∶2)→1.8 min(85∶15)→3.5 min(80∶20)→6 min(75∶25)→7 min(70∶30)→11 min(65∶35)→16 min(0∶100)→26 min(0∶100)→27 min(98∶2)→30 min(98∶2);柱温:40 ℃;进样量:15 μL。
1.2.2 液相条件的优化
1.2.2.1 流动相的选择 通过查阅文献[21-23],在多兽残分析中,普遍采用水-乙腈体系作为流动相,另外添加少量的易挥发酸可提高离子化效率,改善峰性。因此分别考察了0.1%甲酸水溶液-0.1%甲酸乙腈、0.2%甲酸水溶液-0.2%甲酸乙腈、0.1%甲酸-5 mmol/L甲酸铵溶液-0.1%甲酸乙腈这3种流动相对各化合物峰性及响应的影响。
1.2.2.2 色谱柱的选择 为了获得最佳的分离效果和最短的分离时间,考察Thermo Acclaim RSLC C18(100 mm×2.1 mm,2.2 μm)、Agilent ZORBAX Eclipse XDB-C8(50 mm×2.1 mm,1.8 μm)和 Agilent Eclipse Plus C18(150 mm×3.0 mm,1.8 μm)三款色谱柱对待测化合物的分离效果。
1.2.2.3 梯度洗脱的优化 通过调节梯度,优化各化合物的出峰,同时对同分异构体进行分离。存在的三组同分异构体分别为磺胺甲氧哒嗪与磺胺间甲氧嘧啶、磺胺二甲氧嗪与磺胺二甲异噁唑以及吡罗昔康与奥芬达唑砜。
1.2.3 质谱条件 采用电喷雾离子源(Electron Spray Ionization,ESI),正离子模式,检测方式为dMRM,电离电压为3.5 kV,干燥气流速为13 L/min,干燥气温度为150 ℃,鞘气流速为12 L/min,鞘气温度为300 ℃,雾化器流量为35 psi。各化合物质谱参数见表1。
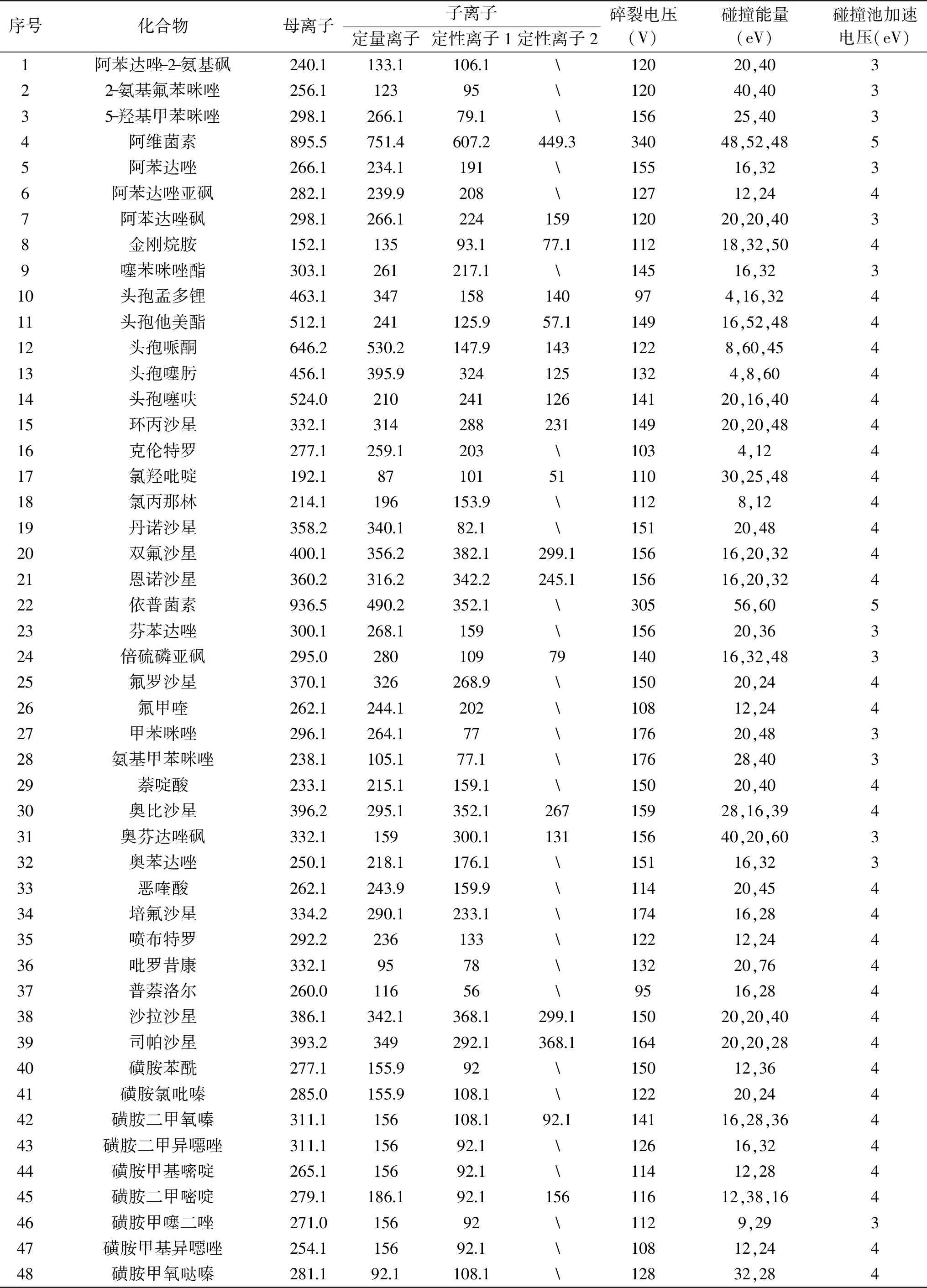
表1 55种化合物质谱参数
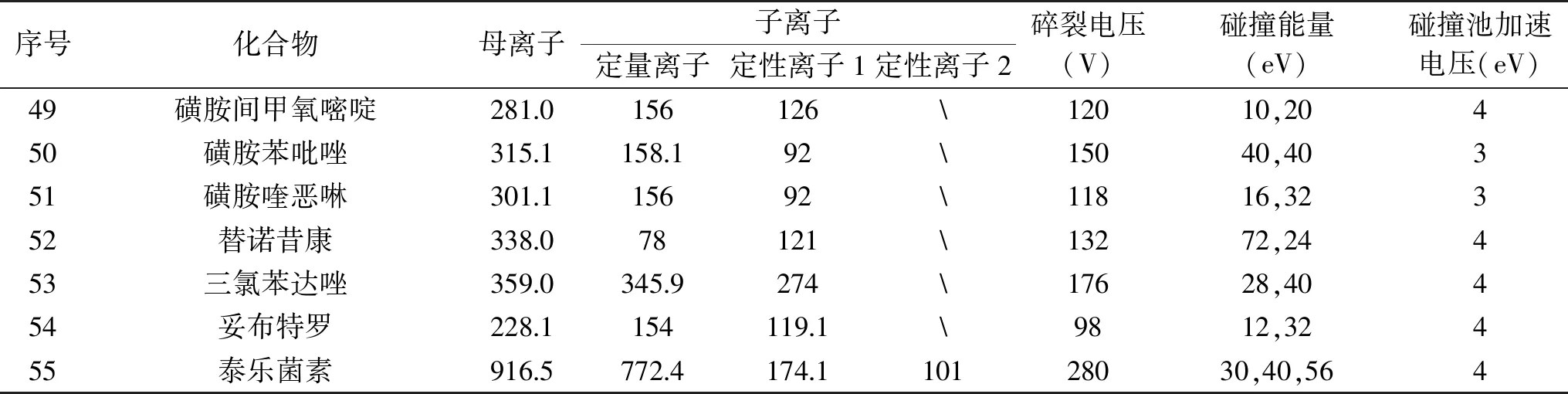
续表
1.2.4 样品前处理
1.2.4.1 提取 称取匀质后的猪肉约5 g,于50 mL具盖塑料离心管中,先加入10.0 mL 提取试剂,再加入两粒陶瓷均质子,涡旋(2000 r/min)混匀 5 min,离心5 min(4000 r/min)。
通过查阅文献[25-27],多兽残分析中多采用乙腈、甲醇等有机试剂作为提取试剂,对高脂肪高蛋白的基质可有效提取目标化合物,降低基质的干扰。与甲醇相比,乙腈提取时带入的极性干扰物更少,有利于后续净化。由于大部分目标化合物多带有含氮基团,具有一定弱碱性,在提取溶剂中适量添加甲酸可提高这类化合物的提取效率。通过实验考察1%、2%、5%、8%甲酸乙腈对55种化合物的提取效率。
1.2.4.2 净化 参考文献[28],移取5.0 mL离心后上层提取液到已活化的EMR-Lipid dSPE管(活化方式:移取5.0 mL 5 mmol/L醋酸铵水溶液到15 mL EMR-Lipid dSPE管中,快速振摇并涡旋2 min(2000 r/min),即可),快速振摇并涡旋2 min(2000 r/min),离心5 min(4000 r/min),将上层液完全倾倒入50 mL离心管中,加入两粒陶瓷均质子及EMR polish粉包,快速剧烈振摇并涡旋2 min(2000 r/min),离心5 min(4000 r/min)。
1.2.4.3 浓缩 精密移取2 mL上层乙腈液至12 mL氮吹管中,加入0.050 mL二甲亚砜,40 ℃下氮气浓缩至约0.050 mL;用15%乙腈水溶液补齐至1 mL,涡旋10 s(2000 r/min),超声10 s,涡旋1 min(2000 r/min);将溶液倒入1.5 mL离心管中,离心2 min(10000 r/min),移取上层液至进样小瓶中,待分析。
1.2.5 方法学验证
1.2.5.1 基质标准曲线的绘制 精密量取55种化合物的标准品各100 μL,置于同一10 mL量瓶中,加乙腈稀释至刻度,作为混合标准品储备液(各化合物浓度为1000 ng/mL)。分别精密移取混合标准品储备液0、5、25、50、100、500、1000 μL至50 mL离心管中(最终浓度分别为0、1.25、6.25、12.5、25、125、250 ng/mL),氮气吹至近干。选取阴性猪肉作为空白基质,分别称取7份(每份5 g),置于上述已加入标准品的离心管中,然后按照样品前处理方法进行操作。以各化合物浓度为横坐标,各化合物定量离子峰面积为纵坐标,绘制基质标准曲线。
1.2.5.2 定量限考察 在确定定量限的研究中,在保证准确定性的前提下,以空白基质将混合标准品储备液分别稀释至10、5、2.5、1.25、0.625 ng/mL来考察定量限。将拟定的定量限通过加标回收的方式考察定性结果、精密度和回收率,在保证定性准确、绝大部分化合物精密度<15%、回收率70%~130%之间的前提下确定定量限。
1.2.6 考察准确性与重复性 以猪肉作为基质,进行添加回收实验。按照1、10、100 μg/kg三水平进行加标回收试验,各水平制样6份。准确性以测出量对理论添加量的百分比值计算平均回收率表示,重复性以测出量的RSD表示。
1.3 数据处理
将获得的数据文件导入Agilent MassHunter Quantitative Analysis(for QQQ)软件进行数据处理。以化合物保留时间作为筛查依据,以离子对丰度比作为确证依据,进行定性判断;通过绘制标准曲线后,将样品中各化合物峰面积带入标准曲线后获得各化合物的浓度,以获得定量结果。
2 结果与分析
2.1 液相条件及优化
2.1.1 流动相的选择 通过比较3种流动相:0.1%甲酸水溶液-0.1%甲酸乙腈、0.2%甲酸水溶液-0.2%甲酸乙腈,0.1%甲酸-5 mmol/L甲酸铵溶液-0.1%甲酸乙腈,其中添加了甲酸铵后部分化合物会出现峰性变差的情况;0.2%甲酸体系中各化合物的响应和分离效果普遍优于0.1%甲酸体系。因此最终采用含0.2%甲酸溶液-0.2%甲酸乙腈作为流动相。
2.1.2 色谱柱的选择 对比三款色谱柱,前两款的分析时间均较短,但由于出峰靠前的化合物(如双氟沙星)峰性变差,而Agilent Eclipse Plus C18对55种化合物均有良好的峰性,因此选其作为分析柱。
2.1.3 梯度洗脱的优化 通过调节梯度,对三组同分异构体进行分离。其中吡罗昔康和奥芬达唑砜产生的二级碎片均不同,不会相互影响定性和定量。但其他两组由于均为磺胺类化合物,会产生相同的碎片。因此在设计梯度洗脱时,需考虑这两组化合物的完全分离。前11 min,选择使用较缓的梯度以使这两组化合物彻底分离。同时,由于55种化合物中44种均在前11 min出峰,11 min后将梯度在5 min之内升到有机相比例100%,并保持10 min,使难洗脱的化合物和基质均能洗脱出来,避免对后续进样的干扰。图1为该条件下总离子流图,可见在此色谱条件下,55种化合物均分离较好。
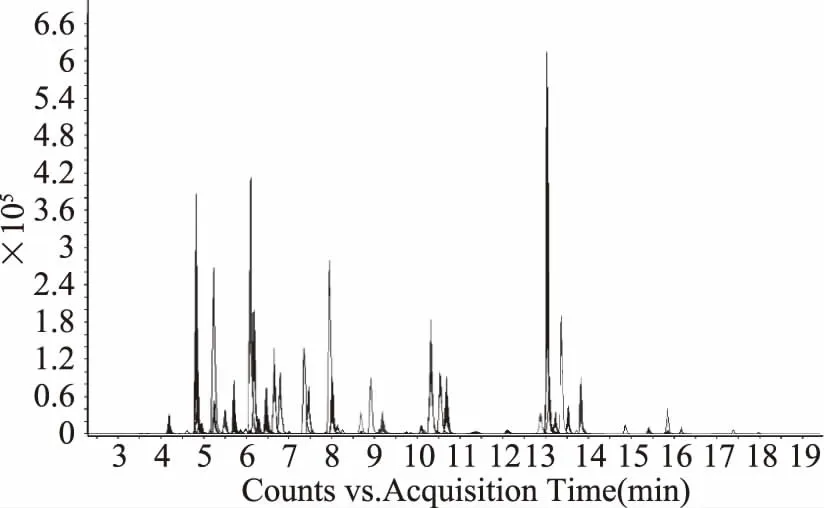
图1 55种化合物总离子流图
2.2 质谱条件
根据欧盟2002/657/EC法案,质谱确证需最低4个识别点数。对于低分辨质谱,母离子的识别点数记为1,每个特征子离子识别点数记为1.5,因此,需至少1个母离子和2个子离子才能满足4个识别点数的要求。由于在实际的样品加标中存在基质对部分化合物定性造成的干扰,包括阿维菌素、阿苯达唑砜、金刚烷胺等20种化合物。因此这部分化合物选择了1个母离子和3个二级子离子以确保定性的准确。

图2 丹诺沙星提取离子流图

图3 丹诺沙星质谱图
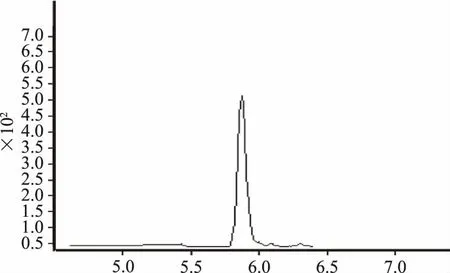
图4 磺胺二甲嘧啶提取离子流图
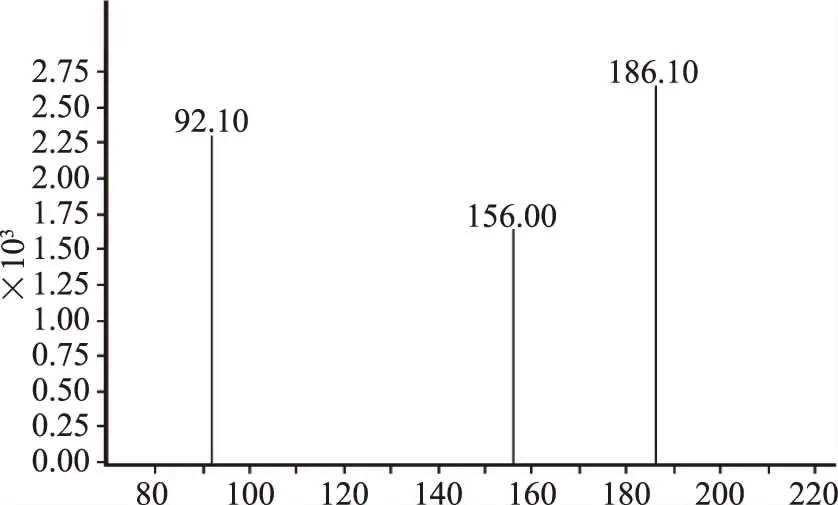
图5 磺胺二甲嘧啶质谱图
2.3 样品前处理
考察1%、2%、5%和8%的甲酸乙腈对各化合物的提取效率。结果发现5%甲酸乙腈作为提取溶剂,大部分化合物回收率均在70%~120%之间。而当酸度较低或过高时,磺胺类化合物回收率均会偏低。
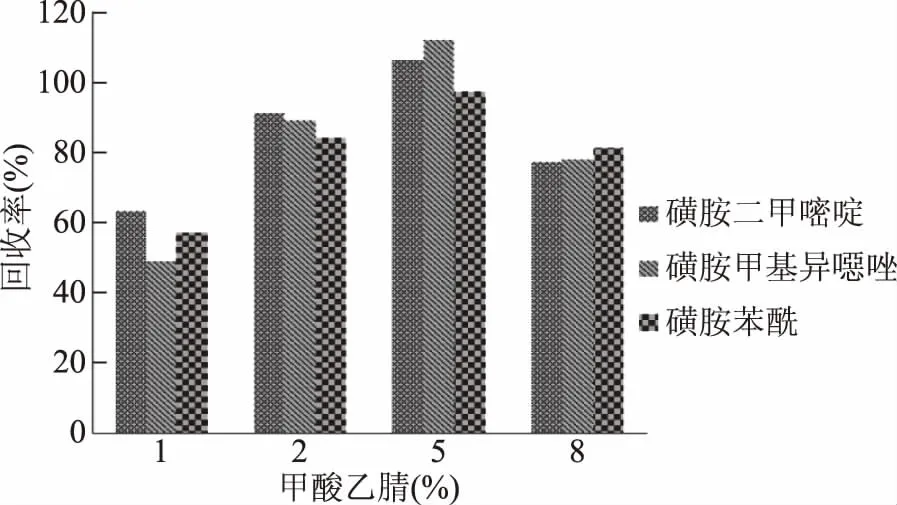
图6 不同酸度对目标化合物回收率的影响
2.4 方法学验证
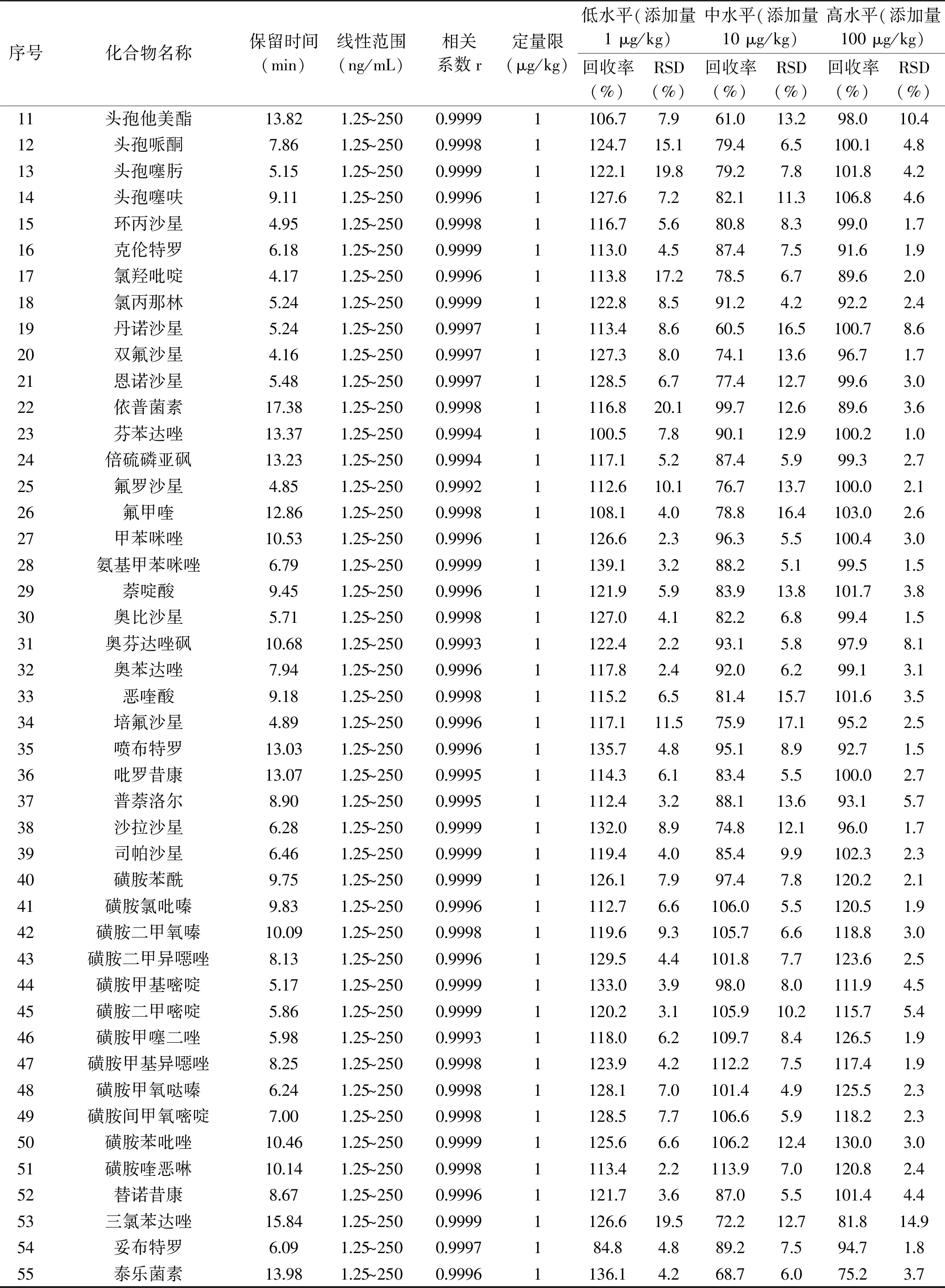
2.4.1 线性与定量限 由线性及定量限结果(见表2)显示,各化合物在1.25~250 ng/mL线性范围内线性良好,相关系数r均≥0.998。定量限均为1 μg/kg。本实验未采用信噪比的方式来确定定量限,原因在于磺胺甲基异噁唑、磺胺甲基嘧啶、氯羟吡啶等化合物在信噪比为10时,定性结果出现假阴性的情况,同时也会出现回收率和精密度不能满足定量分析的要求的情况,比如磺胺甲基嘧啶回收率仅48%,RSD仅32%。

表2 55种化合物线性、定量限、准确度及精密度
续表
2.4.2 准确性与重复性 结果见表2,95%化合物各添加水平的平均回收率为70.0%~130.0%,94%化合物重复性<15%;全部化合物的回收率为60.5%~139.5%,RSD为1.0%~20.1%。方法的准确度和重复性均满足定量分析的要求。
2.5 实际样品测定
在市场上采购猪肉18批次,按照所建立的方法进行测试,结果未发现阳性样品。
3 结论
本文首次将QuEChERS EMR-Lipid与LC/MS/MS联用技术用于猪肉中多兽药残留的快速筛查和确证。采用5%甲酸乙腈作为提取溶剂,经EMR-Lipid净化后,通过Agilent Eclipse Plus C18(150 mm×3.0 mm,1.8 μm)在30 min内完成对55 种化合物的分离,由液质联用仪在正离子及dMRM模式下采集数据,最终由数据处理软件通过保留时间及离子对丰度比进行快速筛查和确证,并采用基质匹配标准曲线定量。所建立的多兽残分析方法,在1.25~250 ng/mL范围内线性良好,相关系数r均≥0.998,定量限均为1 μg/kg,在1~100 μg/kg加标回收中,回收率在60.5%~139.5%,RSD在1.0%~20.1%,准确度和重复性符合定量分析的要求。本方法可实现同时分析55种兽残化合物,大大提高了检验效率,为检验人员提供了一个简单、快捷、高效的检验方法,也为食品安全提供了技术保障。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
核化学与放射化学(2022年2期)2022-04-28
中国饲料(2022年5期)2022-04-26
湖南农业(2020年7期)2020-09-16
智富时代(2019年7期)2019-08-16
智富时代(2019年7期)2019-08-16
钻井液与完井液(2018年2期)2018-06-13
中国蜂业(2018年4期)2018-05-09
中国饲料(2016年5期)2016-02-02
应用化工(2015年1期)2015-12-24
