
高速逆流色谱法分离制备小叶金钱草中杨梅苷与槲皮苷
2019-07-10 13:10宋道光徐顺连樊鑫宇
食品工业科技 2019年11期
宋道光,徐顺连,樊鑫宇,陈 志
(青海师范大学生命科学学院,青藏高原药用动植物资源重点实验室,青海西宁 810008)
小叶金钱草(HydrocotylesibthorpioidesLam.)为伞形科天胡荽属植物天胡荽,中国传统中药之一,为多年生草本植物。小叶金钱草通常生长在海拔475~3000 m湿润的草地、河沟边和林下,广泛分布于中国的许多地区,如四川、广西、贵州等地[1]。目前,小叶金钱草的化学成分含有黄酮类、酚类和香豆素类化合物[2]。超高效液相色谱-串联质谱(UPLC-MS/MS)分析显示小叶金钱草含有酚类和类黄酮,如儿茶素、表儿茶素、槲皮素和绿原酸,并被证明这是小叶金钱草抗氧化活性的主要成分[3],此外,其在体外还具有抗登革热病毒[4]和抗乙型肝炎病毒(HBV)的活性[5]。但截至目前,小叶金钱草只作为民间用药,尚未被中国药典收录,因此对小叶金钱草化学成分及药理活性的深入研究具有重要意义。
虽然传统的硅胶柱层析可以分离出许多化合物,但它需要大量的溶剂,且不能分离具有相似结构和极性的化合物。而高速逆流色谱(high speed counter-current chromatography,HSCCC)是以化合物在两相不互溶的溶剂中分配系数的差异为原理的液液分配色谱,无需固体材料做固定相,无不可逆吸附等作用,具有样品回收率高、成本低、仪器操作简便等特点,近年来在天然产物分离等领域得到了广泛应用[6-7]。
本研究采用高速逆流色谱法分离小叶金钱草中的两种黄酮成分杨梅苷和槲皮苷,有利于进一步研究小叶金钱草的化学成分及其药效,这为小叶金钱草的开发提供理论依据。
1 材料与方法
1.1 材料与仪器
小叶金钱草 青海师范大学植物学陈志教授鉴定,为天胡荽属植物天胡荽,HydrocotylesibthorpioidesLam.),购于西宁城北百信大药房;葡聚糖凝胶(Sephadex LH-20) 安徽博美生物科技有限公司;氯仿、正己烷、乙酸乙酯、正丁醇、乙醇、乙腈、甲酸 分析纯,高效液相色谱所用的乙腈为色谱纯,天津市百世化工有限公司;水 二次蒸馏水。
RE.52AA旋转蒸发器 上海亚荣生化仪器厂;真空脱气过滤装置 美国Waters公司;KJ-11030AL型超声波清洗器 深圳市科洁超声科技有限公司;Waters 600高效液相色谱仪 美国Waters公司;Waters 2998 PAD二极管阵列检测器 美国Waters公司;HSCCC TBE-300B型高速逆流色谱仪(柱体积300 mL) 中国上海同田生化技术有限公司;N2000双通道色谱工作站 浙江大学智达信息工程有限公司;核磁共振仪 瑞士Bruker-500 MHz NMR(AVANCE III HD)。
1.2 实验方法
1.2.1 小叶金钱草乙酸乙酯及分离样品的制备 将购买的小叶金钱草药材用中药粉碎机粉碎,过40目筛。称取20 kg粉末分次置于超声波清洗器,按液料比10∶1加入70%(v:v)乙醇溶液,于600 W、40 kHz、60 ℃提取1 h,过滤除去药渣,将药渣重复提取4次。合并提取液减压浓缩,得到棕褐色粘稠浸膏。浸膏用蒸馏水混悬,依次用10倍量的石油醚、乙酸乙酯、正丁醇萃取5次。取乙酸乙酯萃取液回收溶剂并浓缩得到小叶金钱草乙酸乙酯萃取物,4 ℃避光保存备用。石油醚及正丁醇萃取物的化学成分另做研究。
取5 g乙酸乙酯萃取物,用30 mL蒸馏水超声溶解并过0.45 μm滤膜,Sephadex LH-20柱层析分离(3 cm×80 cm),分别用水、10%、20%、30%、40%、50%、75%、100%甲醇洗脱,每个比例洗脱2个柱体积,1/20柱体积为一馏分,收集75%甲醇洗脱馏分,合并色谱峰保留时间相同的馏分(HPLC检测条件见1.2.2),旋蒸除去溶剂并于-70 ℃冷冻干燥24 h,得到Sephadex LH-20分离样品。其余甲醇洗脱馏分的化学成分另做研究。
1.2.2 分离样品的高效液相色谱检测 取一定量乙酸乙酯+Sephadex LH-20分离样品,用乙腈溶解,0.45 μm针头过滤器过滤后,进行HPLC分析,其中产物的纯度按面积归一化法计算。
HPLC检测条件:色谱柱:Waters 600 SunFireTMC18(4.6 mm×250 mm,5 μm),流动相为乙腈-0.1%甲酸(15%~40%乙腈,0~70 min),流速为1.0 mL/min,柱温为25 ℃,检测波长254 nm。进样10 μL分析。高效液相色谱(HPLC-PAD)紫外光谱扫描范围:200~400 nm。
1.2.3 高速逆流色谱溶剂系统的选择
1.2.3.1 溶剂系统的确定 参考高速逆流色谱分离黄酮类化合物的相关报道选择溶剂系统[8],如乙酸乙酯、正己烷、正丁醇和水等,按照溶剂极性的不同配制比例均为2∶1∶1的乙酸乙酯-正丁醇-水(1号溶剂)和正己烷-正丁醇-水(2号溶剂)系统,考察此两个比例系统对分配系数K的影响。
1.2.3.2 正己烷-正丁醇-水系统比例的确定 按照正己烷-正丁醇-水的体积比配制溶剂系统,比例依次为:1∶1∶1、1∶2∶2、1.5∶1∶1、1.75∶1∶1、1∶2∶1、2∶2∶1和2∶3∶1(3~9号溶剂)。分别考察正己烷和正丁醇对分配系数K的影响。
1.2.3.3 K值的测定 通过高效液相色谱测定目标化合物在溶剂系统中的分配系数K,K值定义为上相中目标化合物的色谱峰面积(S上)除以下相中目标化合物的色谱峰面积(S下)(K=S上/S下)。筛选K值在0.2~5.0的溶剂系统用于高速逆流色谱[9]。另一方面,两组份的分离度(α=K1/K2,K1>K2)应该大于1.5[10]。
分配系数的测定:将适量的样品粉末溶解在适当量预平衡的两相溶剂系统中,并置于分液漏斗中。剧烈摇动分液漏斗以彻底平衡两相样品,并取等体积的两相蒸发至干。然后将残余物用1 mL乙腈溶解过0.45 μm滤膜并按照1.2.2中方法进行HPLC分析。
1.2.4 高速逆流色谱分离制备 配制正己烷-正丁醇-水(1.75∶1∶1,v∶v∶v)两相溶剂系统,于分液漏斗中充分振摇后在室温下静置10 h,将上下相分离并超声脱气20 min,上相为固定相(正己烷、正丁醇)下相为流动相(水)。以20 mL/min的流速将脱气后的上相泵入主机。待主机中充满上相后,启动主机正相旋转按键FWD,调整主机转速为900 r/min,仪器转速稳定后再以2 mL/min的流速泵入超声脱气后的下相。待流动相流出主机后,平衡数分钟待基线平稳后计算固定相保留率(Sf),计算方法为:Sf(%)=[(螺旋管体积-流出固定相体积)/螺旋管体积]×100。
开启TBD 2000检测系统,设置检测波长为254 nm,采集数据。取1.2.1中Sephadex LH-20分离样品50 mg,用10 mL上相液体溶解,由进样阀上样,记录色谱图,收集各分离组分。
1.2.5 化合物核磁共振波谱表征 通过对已分离化合物进行1H NMR和13C NMR谱图的测定,将化合物1H NMR和13C NMR图谱在MestReNova中处理,并对化学位移进行积分和偶和常数(J)的计算。J的计算方法为:(低场化学位移-高场化学位移)×核磁兆数。1H NMR二重峰、双二重峰(分别表示为d及dd)需计算J值,单峰(s)、三重峰(t),以上不计算J值。通过与文献化合物报道数据进行1H NMR化学位移及J和13C NMR化学位移比对,结果一致即确定化合物结构,结构式通过ChemBioDraw 14.0画出。
2 结果与分析
2.1 小叶金钱草乙酸乙酯提取物及Sephadex LH-20分离样品
按照1.2.2方法得到小叶金钱草乙酸乙酯萃取物及Sephadex LH-20分离样品的HPLC色谱图见图1。由图1a可知,小叶金钱草乙酸乙酯萃取物所含化学成分较为复杂,而经Sephadex LH-20纯化后(图1b)HPLC色谱峰较少,有利于分配系数摸索及溶剂系统的优化选择。由图1b可知,峰1、峰2为高速逆流色谱目标分离成分,其保留时间分别为18.974和26.282 min。图1c和图1d分别为峰1和峰2的紫外光谱,色谱峰在峰带I:300~400 nm及峰带II:220~280 nm有明显紫外吸收,符合黄酮类化合物紫外光谱吸收特征,即峰1和峰2为黄酮类化合物。
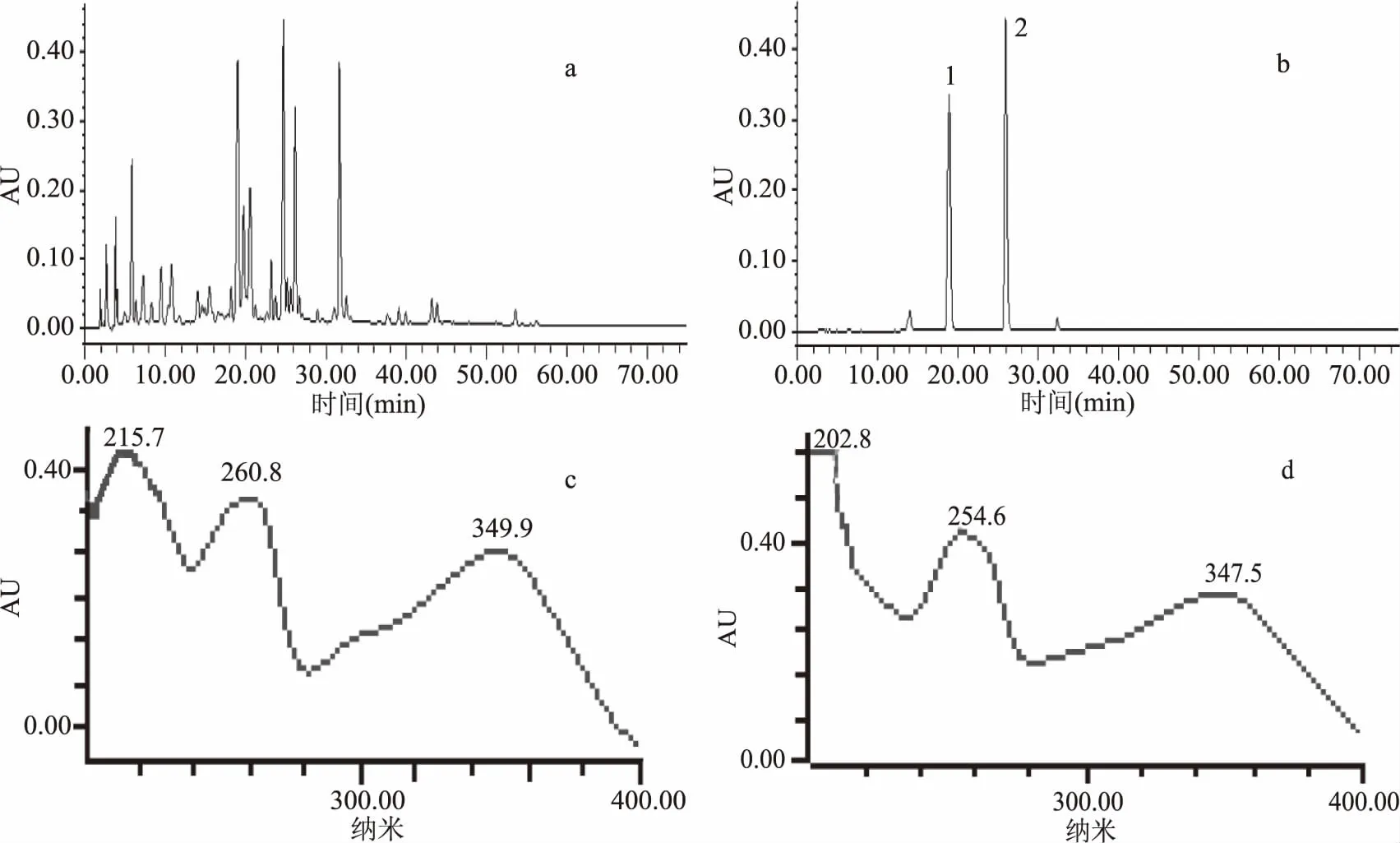
图1 小叶金钱草乙酸乙酯部位(a)、Sephadex LH-20分离样品(b)的高效液相色谱图及峰1(c)、峰2(d)的紫外光谱
2.2 HSCCC分配系数的优化
HSCCC实现好的分离效果的前提为:具有合适分配系数的两相溶剂系统、具有合适的分配系数(0.2~5.0)、两相溶剂分层时间短(小于30 s)和固定相有较高的保留率。
2.2.1 溶剂系统的确定 在初步的试验当中,目标化合物极易溶于乙酸乙酯和水。在表1中可以看出,1号溶剂乙酸乙酯-正丁醇-水(2∶1∶1,v∶v∶v)分配系数过大,因此不适合作为高速逆流色谱溶剂系统。在2号系统中将乙酸乙酯替换为正己烷后体系的分配系数大为改善,但表1中2号系统中1号化合物分配系数较小,会过早地和杂质一同随流动相流出,化合物纯度将无法保证,故不选用2号溶剂系统。因此,下一步通过调整正己烷和正丁醇的比例来探索正己烷-正丁醇-水溶剂系统的最适分配系数。
2.2.2 正己烷-正丁醇-水比例的确定 由表1、表2可知,2号及3~9号溶剂系统的分配系数范围在0.3~11.0之间。在上相作为流动相和下相作为固定相的溶剂体系中,分配系数越小出峰时间越早[11]。通过调整正己烷和正丁醇的比例时可以发现,当增加正己烷或减小正丁醇的比例时,化合物的分配系数减小而分离度增加。由3号及7号、2号及8号和8号及9号可知,增加正丁醇的比例对化合物的分配系数影响较大,即延长了逆流色谱出峰时间。综合考虑,通过调整正己烷的比例可以在保证分离度的前提下尽可能地缩短分离时间,节约溶剂。因此选用6号溶剂系统正己烷-正丁醇-水(1.75∶1∶1,v∶v∶v)进行高速逆流色谱分离目标物。
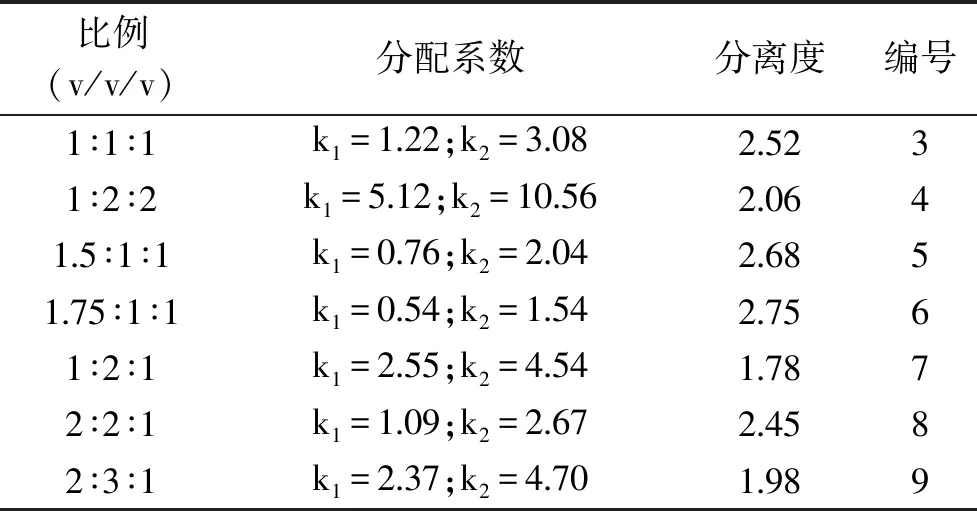
表2 小叶金钱草提取物在正己烷-正丁醇-水溶剂体系中的分配系数 K
2.3 HSCCC分离结果
采用溶剂系统正己烷-正丁醇-水(1.75∶1∶1,v∶v∶v),参照1.2.3中固定相保留率计算方法,Sf=[(300-104)]/300×100%=65.3%。按照1.2.4的方法进行高速逆流色谱分离,得到3个分离组分(图2)并进行HPLC分析。峰I为混乱杂质峰,没有进一步分离价值。峰Ⅱ~Ⅲ样品HPLC分析结果见图3,其中峰Ⅱ图3a、峰III图3b为目标化合物,保留时间(Rt)和纯度分别为:峰Ⅱ,Rt=18.974 min,97.85%;峰Ⅲ,Rt=26.282 min,95.42%。图3中小图对应各色谱峰的紫外吸收光谱,由紫外光谱分析得知,色谱峰紫外吸收在峰带I:300~400 nm及峰带Ⅱ:220~280 nm有明显紫外吸收,符合黄酮类化合物紫外光谱吸收特征,故所分离得到的两种化合物为黄酮类化合物。样品冷冻干燥后得峰Ⅱ 18.63 mg,峰Ⅲ 17.49 mg。
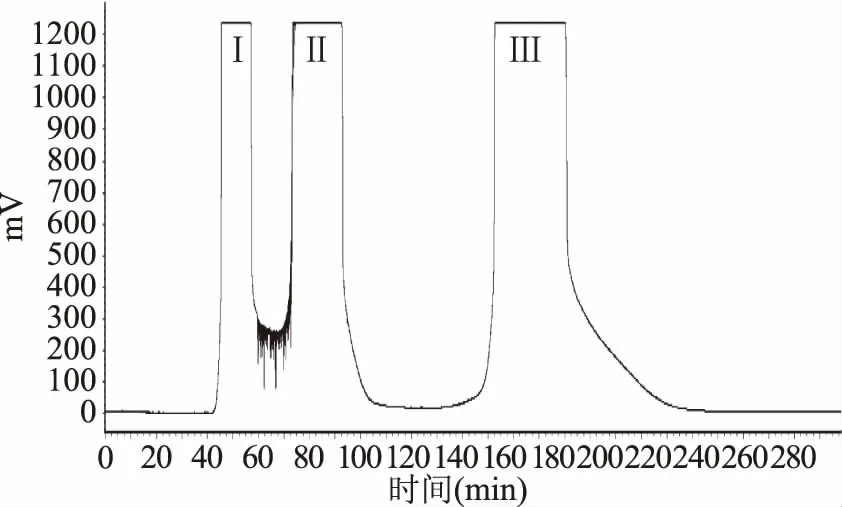
图2 小叶金钱草乙酸乙酯部位分离样品的高速逆流色谱图
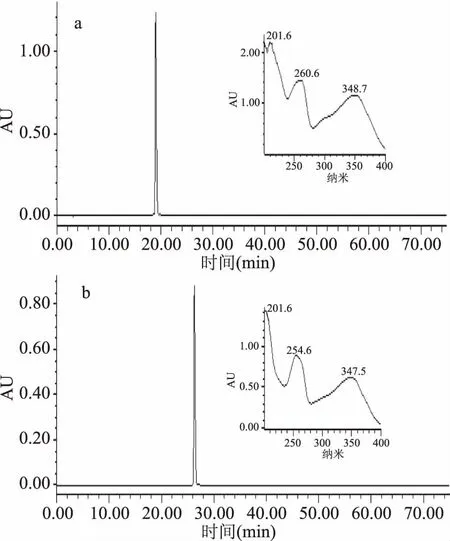
图3 峰Ⅱ(a)、Ⅲ(b)组分的 HPLC色谱图
2.4 结构表征
化合物1的1H NMR和13C NMR数据如下:1H NMR(DMSO-d6,500 MHz,ppm):δ 12.69(1H,s,5-OH),9.26(4H,br s,3′,4′,5′,7-OH),6.90(2H,s,2′,6′-H),6.37(1H,d,J=1.9 Hz,8-H),6.20(1H,d,J=1.8 Hz,6-H),5.12(1H,s,1″-H),3.15-3.99(4H,m,rha-2,3,4,5),0.85(3H,d,J=6.2 Hz,CH3);13C-NMR(DMSO-d6,125 MHz,ppm):δ 177.2(C-4),164.9(C-7),161.8(C-5),157.9(C-8a),156.9(C-2),146.25(C-3′,5′),136.9(C-4′),134.7(C-3),120.0(C-1′),108.4(C-2′,6′),104.4(C-4a),102.4(C-1″),99.2(C-6),94.0(C-8),71.7(C-4″),71.0(C-3″),70.9(C-2″),70.5(C-5″),18.0(C-6″)。根据已报道文献比对[12],化合物1为杨梅苷(Myricitrin),其结构式见图4(1)。1H NMR和13C NMR图谱分别见图5a和图5b。

图4 杨梅苷(1)和槲皮苷(2)的化学结构

图5 杨梅苷(a,b)和槲皮苷(c,d)的核磁共振氢谱碳谱(500 MHz,DMSO-d6)
化合物2的1H NMR和13C NMR数据如下:1H NMR(DMSO-d6,500 MHz,ppm):δ 12.66(1H,s,5-OH),9.74(3H,br s,7,3′,4′-OH),7.31(1H,d,J=2.0 Hz,2′-H),7.26(1H,dd,J=2.0 Hz 和 J=8.3 Hz,6′-H),6.87(1H,d,J=8.3 Hz,5′-H),6.39(1H,d,J=1.7 Hz,8-H),6.20(1H,d,J=1.8 Hz,6-H),5.27(1H,s,1″-H),3.14-3.99(4H,m,rha-2,3,4,5),0.83(3H,d,J=6.1 Hz,CH3);13C-NMR(DMSO-d6,125 MHz,ppm):δ 178. 2(C-4),165.1(C-7),161.8(C-5),157.7(C-2),157.0(C-8a),148.9(C-4′),145.7(C-3′),134.6(C-3),121.6(C-6′),121.2(C-1′),116.1(C-5′),116.0(C-2′),104.4(C-4a),102.3(C-1″),99. 3(C-6),94.2(C-8),71.6(C-4″),71.1(C-2″),70.8(C-3″),70.5(C-5″),17.9(C-6″);根据已报道文献比对[13],化合物2槲皮苷(Quercitrin),其结构式见图4(2)。1H NMR和13C NMR图谱分别见图5c和图5d。
3 结论
本文采用高速逆流色谱法首次从小叶金钱草中分离制备两种黄酮苷类化合物,确定两相溶剂系统为正己烷-正丁醇-水(1.75∶1∶1,v∶v∶v),分配系数达到0.54和1.54,经1H NMR和13C NMR确定此两种物质分别为杨梅苷和槲皮苷,其纯度分别达到97.85%和95.42%。经实验研究建立从小叶金钱草分离纯化杨梅苷和槲皮苷的HSCCC方法,为进一步研究小叶金钱草的化学成分、药效以及综合利用奠定了一定的理论基础。
猜你喜欢
承德医学院学报(2022年2期)2022-05-23
今日中国·西班牙文版(2020年11期)2020-11-06
小学生作文(低年级适用)(2019年4期)2019-04-29
中成药(2018年10期)2018-10-26
中成药(2018年9期)2018-10-09
中成药(2018年7期)2018-08-04
中成药(2018年3期)2018-05-07
天然产物研究与开发(2018年3期)2018-05-07
散文诗(2017年18期)2018-01-31
家庭科学·新健康(2017年7期)2017-07-14
