
葛根素与水二聚体相互作用的密度泛函理论研究
2019-06-25 01:55黄茂胜黄嗣航陈求芳
中国民族民间医药·下半月 2019年2期
黄茂胜 黄嗣航 陈求芳
【摘 要】 采用DET-B3LYP/6-31+G方法优化了葛根素与水的二聚体几何结构和电子结构,计算二聚体的几何构型,电荷分布及转移,结合能和热力学性质。探讨葛根素与水分子之间相互作用的结构及能量变化,旨在密度泛函理论上寻找其最稳定结构。结果显示,与单体相比,各二聚体在接触点上的氧原子,氢原子及附近的氧原子变化明显,两子体系间的电荷转移较多。二聚体(Ⅰ~Ⅵ)中的接触点(O-H…O)距离最小为0.1580nm,最大的结合能和焓变值分别为79.82kJ·mol-1和-20.92kJ·mol-1均属于构型Ⅲ。即葛根素与水相互作用形成二聚体(Ⅰ~Ⅵ)中,二聚体Ⅲ为最稳定结构。
【关键词】 葛根素;水;二聚体;密度泛函理论;相互作用
【中图分类号】R285.5 【文献标志码】 A【文章编号】1007-8517(2019)4-0041-04
Abstract:The optimized geometries and electronic structures of puerarin, water and their possible stable dimers were obtained at the DFT-B3LYP/6-31+G level. Structures, thermodynamics, energy and charge were also measured. To investigate the change to the structure and energy between puerarin and water molecules as well as its optimal structure by density functional theory. Compared with the monomer, charge redistribution mainly occurred on the adjacent O-H…O atoms of puerarin dimers between submolecules while the change of the charge between two submolecules was large for all two Puerarin-water Dimers. In addition, the minimum distances of the connection point(O-H…O) in Dimers(Ⅰ~Ⅵ) that belong to the dimer Ⅲ reached 0.1580nm. The maximum corrected binding energy and enthalpy of Puerarin-water Dimers that also belong to the dimer Ⅲreached 79.82kJ·mol-1, -20.92kJ·mol-1. The DimerⅢ is the most stable in the constitution of Puerarin-water Dimers(Ⅰ~Ⅵ).
Keywords:Puerarin;Water;Dimer;Density functional theory;Interaction
葛根素(Puerarin)来源于豆科植物野葛(Puerarin Lobata(Willd.)Ohwi)或甘葛藤(Puerarin thomsonii Benth.)的干燥根中提取出的一种异黄酮苷[1],分子式为C21H20O9,相对分子质量为416.38,呈白色针状结晶。其毒性小、安全范围广、疗效好,临床上主要用于心脑血管疾病的治疗[2]。但其水溶性较差限制了葛根素在临床上的广泛使用[3]。葛根素具有异黄酮类物质化学结构的特点,β环受吡喃环羰基的立体阻碍影响,在空间上只能形成大的共扼体系而成为非平面结构,且晶格排列紧密,刚性较强[4]。葛根素在8位上连一吡喃葡萄糖,在7,4位上各有一羟基,为葛根素的活性基团。其苷元水溶性很差,因此认为葡萄糖基是葛根素有一定水溶性的重要因素。而结构中的羟基(-OH)均有可能和水分子形成分子间氢键,所以本研究设计了6种C21H20O9-H2O超分子体系的结构,通过比较分子之间的相互作用强弱,来揭示相互作用的本质,对了解葛根素与水的作用方式有重要意义[5]。
1 计算方法
分子模拟的主要方法有经典力学模拟和量子力学模拟,而Gaussian一般适用于量子力学计算。其中量子力学模拟主要依据的方法有从头计算法、半经验分子轨道理论、密度泛函理论(DFT)方法等。而密度泛函方法是一个较好的理论计算和结构预测方法,对于确定复杂结构具有重要的參考意义[6]。本文以Gview 3.7软件组建葛根素单体C21H20O9和H2O的二聚体C21H20O9-H2O,计算方法均使用DET-B3LYP/6-31+G方法。其中,B3LYP是密度泛函理论中常用的方法之一,6-31G是描述原子的基组,“6”是指对内层轨道的描述;“31”是指每个价层电子会被劈裂成2个基函数,分别由3个和1个高斯型函数线性组合而成;“G”是高斯基组。以本征值跟踪算法(EF)和内坐标的Berny算法等[7]对所选的结构进行全优化,求得能量极小点(稳定构型),经振动分析得优化结构无虚频。采用Boys和Bernardi提出的均衡校正法(CP)[8]进行基组叠加误差(BSSE)校正与零点能(ZPE)校正。通过自然轨道分析[9],揭示单体转变为二聚体后的电荷转移状况,探索C21H20O9和H2O形成二聚体时的相互作用的本质。通过统计热力学方法求得热力学函数[10],揭示单体转变为二聚体后的能量变化,探索C21H20O9在溶解过程的变化规律及反应机制。全部计算均采用Guassian 09W程序[11]完成,收敛精度取程序内定值。
2 结果与讨论
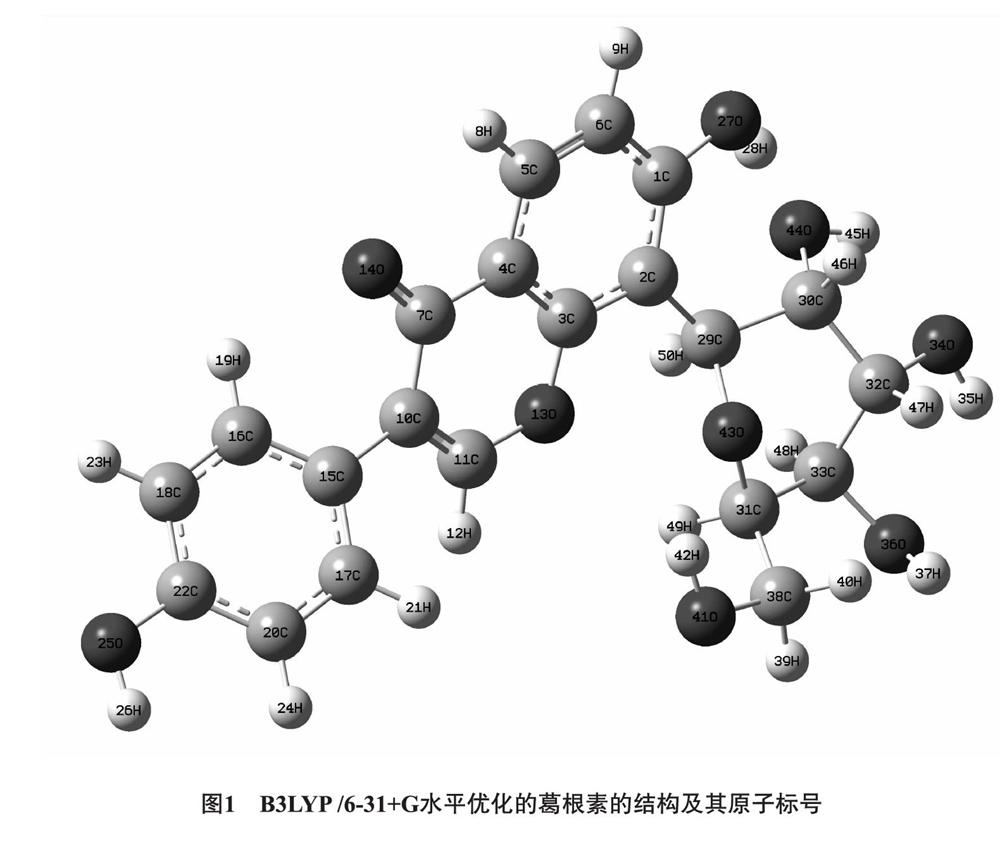
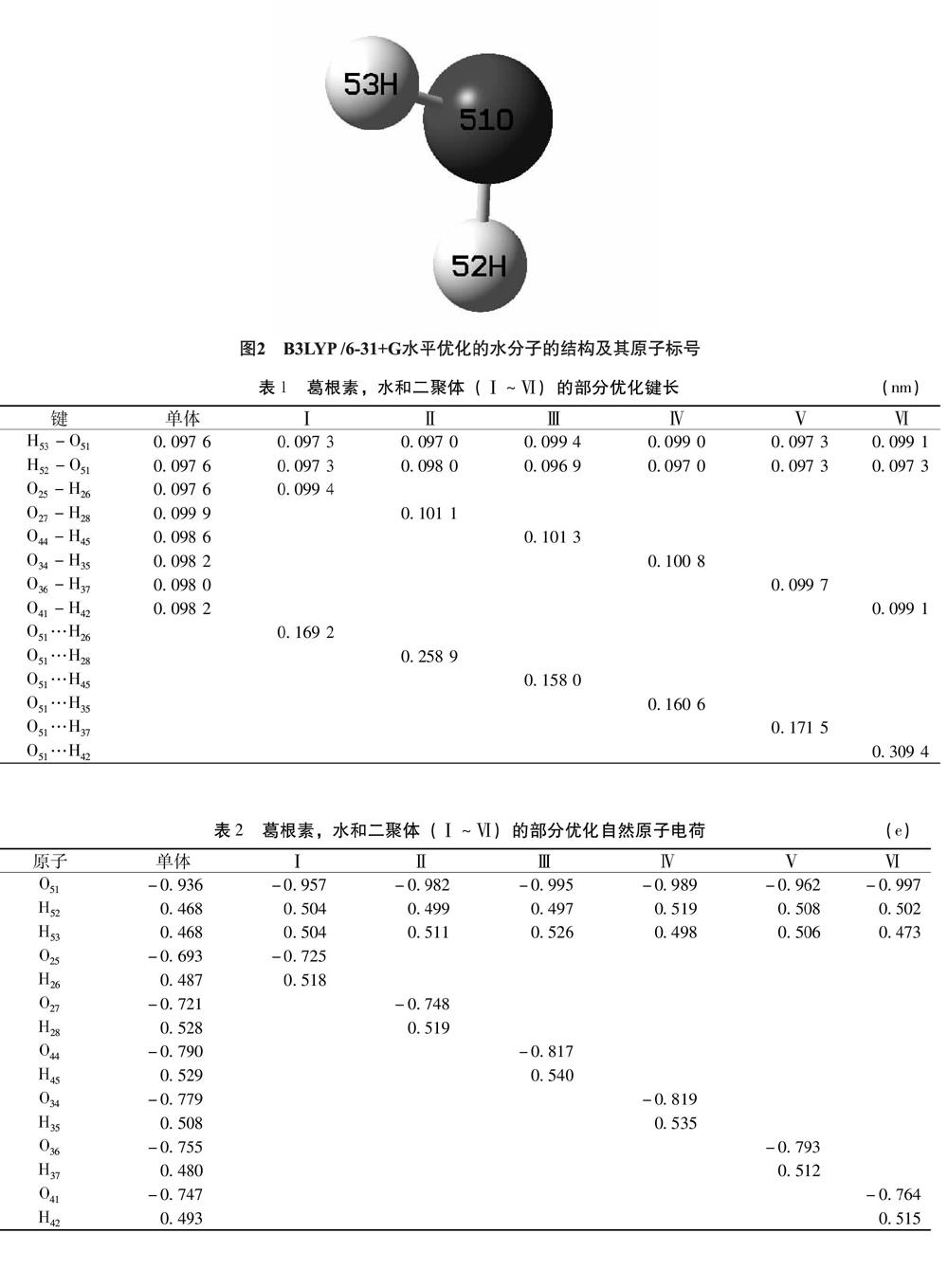
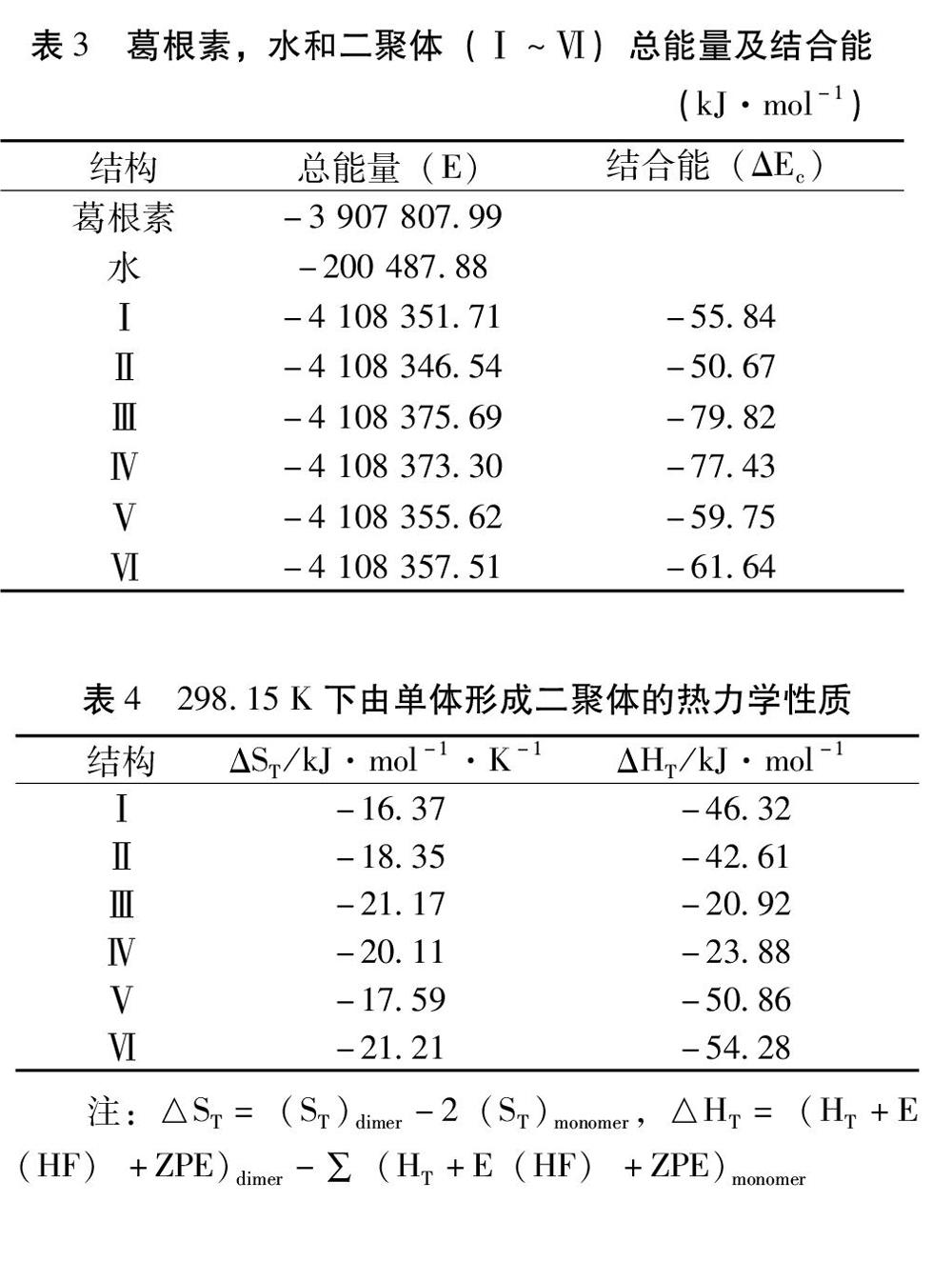
2.1 几何构型 图1和图2列出了优化后的葛根素和水分子的及原子编号,二聚体(Ⅰ~Ⅵ)的DFT-B3LYP/6-31+G全优化几何构型部分几何参数列于表1。其中酚羟基O25-H26(即葛根素异黄酮母核B环上的酚羟基)与水形成的二聚体称为二聚体Ⅰ;酚羟基O27-H28(即葛根素异黄酮母核A环羟基)与水形成的二聚体称为二聚体Ⅱ;羟基O44-H45与水形成的二聚体称为二聚体Ⅲ;羟基O34-H35与水形成的二聚体称为二聚体Ⅳ;羟基O36-H37与水形成的二聚体Ⅴ;羟基O41-H42(即葡萄糖五元环结构所连亚甲基上的羟基)于水形成的二聚体称为二聚体Ⅵ。
由表1可见,二聚体(Ⅰ~Ⅵ)中的接触点(O-H…O)距离分别为0.1692、0.2589、0.1580、0.1606、0.1715和0.3094nm,长短次序为:Ⅲ<Ⅳ<Ⅰ<Ⅴ<Ⅱ<Ⅵ,接触点距离最短属于二聚体Ⅲ。二聚体(Ⅰ~Ⅵ)均近似地属于C1点群,与单体相比,二聚体(Ⅰ~Ⅵ)接触点为O-H…O的O-H键长变化分别为0.0018、0.0012、0.0027、0.0026、0.0017和0.0009nm,二聚体Ⅲ的O-H键长变化最大。与单体相比,二聚体键角变化均小于0.8°,二面角变化均小于0.2°。由此可见,聚合和分子间相互作用未造成各单体较大的扭曲,对内旋转影响也比较小。根据二聚体的接触点种类和距离可以初步推测出结果:二聚体Ⅲ的稳定性最优。
2.2 电荷分布及转移 由表2可见,与单体相比,在接触点上的氧原子,氢原子及附近的氧原子变化明显。如二聚体Ⅲ中的O44为-0.817e减少了0.027e,H45为0.540e增加了0.011e。结合后H2O上的氧原子和氢原子的电荷也有明显变化。子体系间电荷转移比较多。在二聚体(Ⅰ~Ⅵ)中两子体系间电荷传递主要通过接触点O-H…O传递电荷分别为0.051、0.028、0.029、0.028、0.052和0.022e。
2.3 结合能 由表3可知,在DFT-B3LYP/6-31+G水平下求得的葛根素和水形成二聚体时的分子间相互作用能,有总能量(E)和经基组叠加误差(BSSE)和ZPE校正后的结合能(ΔEc)两部分。二聚体(Ⅰ~Ⅵ)结合能分别大小顺序为:Ⅲ﹥Ⅳ﹥Ⅵ﹥Ⅴ﹥Ⅰ﹥Ⅱ。经BSSE和ZPE校正后的葛根素与水二聚体的最大结合能为-79.82kJ·mol-1,属于构型Ⅲ,而二聚体Ⅱ的结合能较低,这是由于所连接的酚羟基受葡萄糖糖基的位阻影响,活性较弱,这与前面的稳定化能及根据键长和接触点距离来推测二聚体稳定性基本相一致。
2.4 热力学性质 基于统计热力学的方法,在振动分析基础上(频率校正因子为0.96[10]),计算在298.15K下由单体生成二聚体(Ⅰ~Ⅵ)的热力学函数的变化值,见表4。由单体生成二聚体时,体系熵值减小,分子间相互作用使聚合过程焓值变大,是吸热过程,形成二聚体(Ⅰ~Ⅵ)焓变值Ⅲ﹥Ⅳ﹥Ⅴ﹥Ⅵ﹥Ⅱ﹥Ⅰ,表明二聚体Ⅲ,Ⅳ,Ⅴ,Ⅵ,Ⅱ,Ⅰ分子间相互作用依次减弱。分子间相互作用最强的二聚体为Ⅲ,这与前面推论一致。
3 结论
由葛根素结构分析,黄酮母核上的有两个酚羟基,由于酚羟基与苯环已形成了共轭体系,很难像醇羟基一样发生反应,故酚羟基上与水分子发生氢键作用形成二聚体的稳定性可能较差。因而连接在葡萄糖苷上的4个羟基与水相互作用形成的二聚体稳定性可能较优。
在DFT-B3LYP/6-31+G水平下,求得的葛根素与水二聚体的优化结构(Ⅰ~Ⅵ)。而且经振动分析得优化结构无虚频,具有稳定的结构。与单体相比,二聚体(Ⅰ~Ⅵ)接触点为O-H…O的O-H键长均变大,接触点距离最小的属于构型Ⅲ。而且各接触点上的氧原子,氢原子及附近的氧原子电荷变化明显。对葛根素与水形成二聚体(Ⅰ~Ⅵ)的自然键轨道,相互作用能和热力学性质上来分析,最大的结合能和焓变值分别为79.82kJ·mol-1和-20.92kJ·mol-1,均属于构型Ⅲ。因此,二聚体Ⅲ为最稳定结构。
参考文献
[1]张彬, 向纪明. 葛根素结构修饰的研究进展[J]. 安康学院学报, 2015, 27(5):99-103.
[2]吴晶, 高星, 张瑜,等. 葛根素结构改造研究进展[J]. 中国药物警戒, 2011, 8(12):741-743.
[3]郭波红, 许丹翘, 孙树铭,等. 表面活性剂及高分子物质对葛根素溶解特性的影响[J]. 亚太传统医药, 2016, 12(17):14-17.
[4]王劲, 周长征. 葛根素衍生物的合成研究进展[J]. 山东中医药大学学报, 2011(4):369-371.
[5]Xing Z H, He L J, Sun Z W, et al. Effect of pH value of solution on stability of puerarin structure[J]. Journal of Harbin University of Commerce, 2015(6):650-652
[6]徐为人, 刘成卜, 王建武. 四氢小檗碱的三维结构和核磁共振的理论研究[J]. 中草药, 2003, 34(12):1068-1071.
[7]Mench M M, Kuo K K, Yeh C L, et al. Comparison of thermal behavior of regular and ultra-fine aluminum powders (Alex) made from plasma explosion process[J]. Combust. Sci. Technol, 1998, 135(1):269-292.
[8]黄辉, 黄勇, 李尚斌. 含纳米级铝粉的复合炸药研究[J]. 火炸药学报, 2002, 25(2):1-3.
[9]Fathauer R W, George T, Ksendzov A, et al. Visible luminescence from silicon wafers subjected to stain etches[J]. Applied Physics Letters, 1992, 60(8):995-997.
[10]Pople J A, Schlegel H B, Krishnan R, et al. Molecular orbital studies of vibrational frequencies[J]. International Journal of Quantum Chemistry, 2009, 20(s):269-278.
[11]Yu W F, Huang H, Nie F D, et al. Research on Nano-Composite Energetic Materials[J]. Energetic Materials, 2005,13(5):340-344.
(收稿日期:2018-12-12 編辑:刘斌)
猜你喜欢
昆明医科大学学报(2021年4期)2021-07-23
中成药(2018年6期)2018-07-11
中成药(2017年6期)2017-06-13
分析化学(2016年7期)2016-12-08
考试周刊(2016年85期)2016-11-11
中国卫生标准管理(2015年16期)2016-01-20
湖北理工学院学报(2015年1期)2015-02-27
现代检验医学杂志(2015年2期)2015-02-06
现代检验医学杂志(2015年1期)2015-02-06
现代检验医学杂志(2014年5期)2014-02-02
