
HPLC-PAD法测定硫酸新霉素含量及其有关物质
2019-05-23 02:14黄敏文FANGWang杭太俊袁耀佐
中国药科大学学报 2019年2期
张 倩,黄敏文,FANG Wang,张 玫,杭太俊,袁耀佐**
(1江苏省食品药品监督检验研究院,南京 210019;2中国药科大学药物分析教研室,南京 210019;3St Jude Children′s Research Hospital,Memphis,TN,USA)
硫酸新霉素(neomycin sulfate)是1949年从新霉素链霉菌(streptomyces fradiae)的代谢产物中分离得到的混合物[1](图1),含有A、B、C 3个成分,其中新霉素B和C为主要成分,新霉素A(即新霉胺)仅微量。其主要通过与细菌核糖体30S亚单位结合,抑制细菌蛋白质的合成[2-3]。新霉素的耳、肾毒性较强,但当局部应用时,很少引起毒性或过敏性反应,因此主要用于治疗皮肤感染性疾病;新霉素口服极少吸收,在肠道内浓度很高,与其他抗生素混合口服可用于对肠道微生物群系的控制,适合于手术前的肠道消毒等[4-5]。其原料药在《中国药典》(2015)(ChP2015)、欧洲药典9.0(EP9.0)、国际药典第五版(Ph.In 5th)、美国药典40-国家处方集35(USP40-NF35)、日本药局方17版(JP17)、韩国药典第10版均有收录[6-11],含量测定均采用专属性差、繁琐耗时的抗生素微生物检定法,但有关物质检查项除EP9.0外,其他药典中均未设定。

Figure1 Chemical structures of neomycin B,neomycin C and neamine
脉冲安培电化学检测器(pulsed amperometric detector,PAD)是通过测定电化学活性物质在工作电极表面发生氧化或还原反应时所产生的电流变化实现对其定量分析,其与HPLC联用在氨基糖苷类抗生素质量控制中已有很多应用,因其灵敏度高、专属性好、浓度与响应呈线性等优点越来越受到重视,并有多个品种被相关药典收载[12],在ChP2015版的硫酸依替米星原料药的标准中,也首次将该技术应用于产品质量控制。
EP9.0采用高效液相色谱-脉冲安培检测法测定硫酸新霉素的有关物质,但在预实验过程中发现,该方法还有一些不足之处。EP9.0采用三电位检测波形(triple-potential waveform,TPW)施加电位的检测模式,文献报道[13-15]及实践表明,TPW模式存在电极耗损快,方法重复性、重复性及耐用性差等缺点,而四电位检测波形(quadruple-potential waveform,QPW)能较好改善上述不足,已成为氨基糖苷类抗生素质量控制的新手段,并在阿米卡星、依替米星等检测时得到应用。另外,在EP9.0色谱条件下,原料中的部分杂质与主峰及各杂质之间的分离欠佳。
因此,本研究在EP9.0方法的基础上,采用QPW作为检测波形,对色谱条件进行优化,并进行了系统的方法学验证,建立了HPLC-PAD法测定硫酸新霉素的含量和有关物质的方法,与EP9.0方法相比,该方法在专属性、灵敏度和重复性等方面均取得较大改善。
1 材 料
1.1 仪 器
Dionex ICS-5000+离子色谱仪(美国Dionex公司);脉冲安培电化学检测器(Ag-AgCl参比电极;金电极,3mm);变色龙数据分析软件(Chromeleon7.2 SR4);PC10池(Pneumatic Controller);XS205电子天平(瑞士Mettler Toledo公司)及Milli-Q IQ7000超纯水发生装置(美国Millipore公司)。
1.2 药品与试剂
新霉素标准品(批号:130309-201512)、巴龙霉素(批号:130371-200305)、新霉胺标准品(批号:130411-200908)均来源于中国食品药品监督检验研究院。硫酸新霉素B精制品(以新霉素B计:66.007%,批号:180803)、硫酸新霉素C精制品(以新霉素C计:60.776%,批号:180801,宜昌三峡制药有限公司);新霉胺精制品(96.2%,无锡福祈制药有限公司)。12批硫酸新霉素原料药来源于A、B两个不同企业2018年国家评价性抽验供研究用样品。
五氟丙酸、50%无二氧化碳氢氧化钠水溶液(HPLC级,美国Sigma-Aldrich公司);三氟乙酸(HPLC级,J&K Scientific公司);高纯氦气(南京天泽气体有限责任公司);超纯水。
2 方 法
2.1 色谱条件
色谱柱:Thermo AcclaimTMAmG (4.6 mm×150mm,3 μm);以2%三氟乙酸溶液[含0.01%五氟丙酸+0.6%氢氧化钠]为流动相;流速:0.8 mL/min;柱温:25 ℃;进样量:25 μL。用积分脉冲安培电化学检测器检测,检测电极为金电极(推荐使用3 mm直径),参比电极为Ag/AgCl复合电极,钛合金对电极,四波形检测电位(图2,0.2~0.4 s检测信号积分),柱后碱液:50%NaOH溶液(1.0 mL 50% NaOH加水制成25 mL溶液),碱液推荐流速:0.3 mL/min。
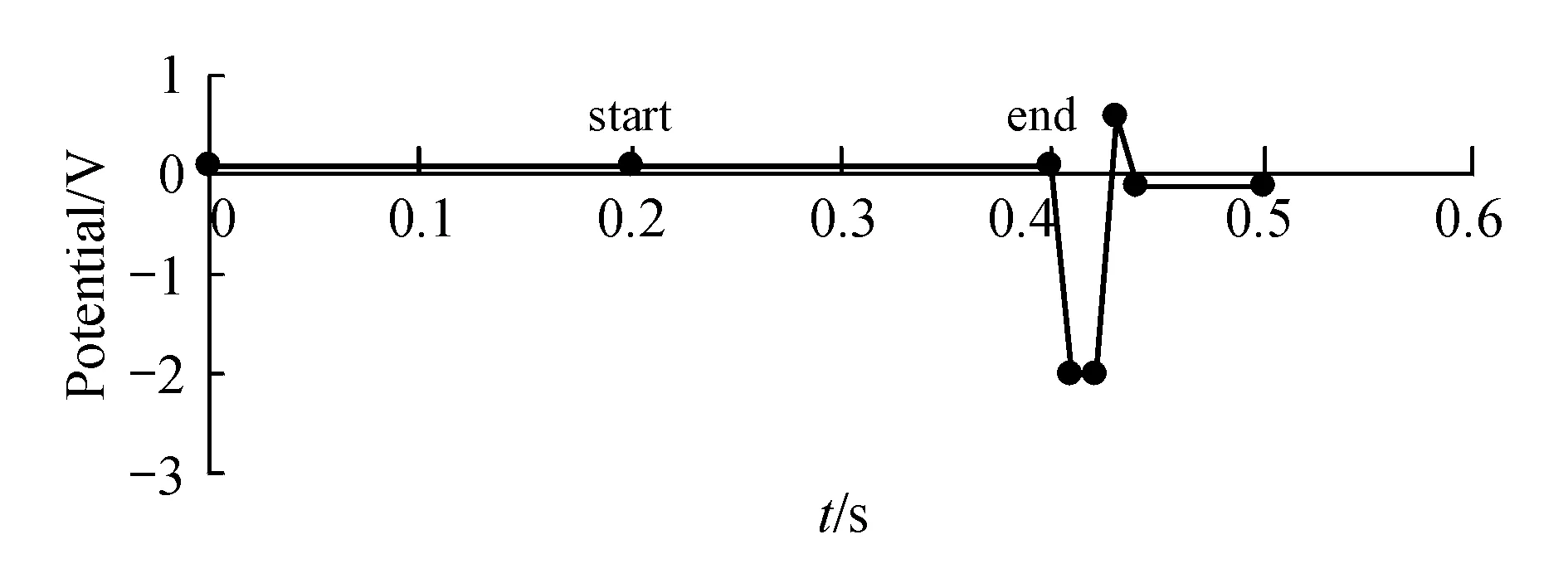
Figure2 Settings of quadruple-potential waveform-time
2.2 溶液的制备
2.2.1 柱后碱液 量取超纯水960 mL,以50 mL/min流量通氦气脱气8 min后,立即量取质量分数为50%的氢氧化钠溶液40 mL混合,继续脱气3 min,即得。
2.2.2 系统适用性溶液 精密称取新霉胺精制品及新霉素标准品,加水溶解并稀释成每毫升含新霉胺10 μg、硫酸新霉素30 μg的溶液,即得。
2.2.3 标准溶液 精密称取硫酸新霉素标准品(标准品采用硫酸新霉素B精制品及硫酸新霉素C精制品标化),加水溶解并稀释成含新霉素B分别为0.14,0.34,0.69,1.38,3.4,6.89,13.79,27.89 μg/mL的溶液,及含新霉素C分别为0.14,0.28,0.70,1.41,2.82,4.69 μg/mL的溶液,精密称取新霉胺精制品,加水溶解并稀释成含新霉胺分别为0.36,0.76,1.51,3.03,6.06,12.11 μg/mL的溶液。
2.2.4 含量测定溶液 精密称取硫酸新霉素供试品和新霉素标准品适量,加水溶解并稀释制成30 μg/mL的溶液,分别作为供试品溶液和对照溶液。
2.2.5 有关物质测定溶液 精密称定硫酸新霉素供试品,用流动相溶解并稀释制成0.4 mg/mL的溶液为供试品溶液,精密量取2 mL,用流动相稀释制成8 μg/mL的溶液为对照溶液。
3 方法学验证
3.1 系统适用性
取“2.2.2”项系统适用性溶液,按照“2.1”色谱条件下测定,出峰顺序依次为:新霉胺、新霉素C及新霉素B。新霉素B、新霉素C与相邻杂质的分离度均符合规定。
3.2 专属性
取硫酸新霉素原料药,加水稀释成含硫酸新霉素0.4 mg/mL的溶液,作为供试品溶液。取上述溶液,分别经酸破坏(0.1 mol/L 三氟乙酸破坏10 min后,0.1 mol/L NaOH中和)、碱破坏(0.1 mol/L NaOH破坏10min后,0.1 mol/L 三氟乙酸中和)、氧化破坏(30% H2O2密闭环境放置1 h)、高温(60 ℃、80 ℃水浴中水浴1 h)和光照破坏(254 nm紫外照射20 h)处理,结果见图3。
结果表明,通过酸、碱、氧化、高温、紫外光照射处理样品后,照“2.1”项色谱条件下,主峰与杂质之间,以及杂质与杂质之间分离良好,在酸破坏条件下,新霉胺峰面积略有增加,表明该方法的专属性良好。
3.3 线性及新霉胺校正因子
新霉素B:在0.14~27.89 μg/mL的质量浓度范围内线性关系良好,线性回归方程为Y=3.628 3X+1.952 6,r=0.998 5。
新霉素C:在0.14~4.69 μg/mL的质量浓度范围内线性关系良好,线性回归方程为Y=3.280 9X+0.081 4,r=0.999 6。
新霉胺:在0.36~12.11 μg/mL的质量浓度范围内线性关系良好,线性回归方程为Y=2.662 7X+1.405 2,r=0.998 0。
新霉素C对新霉素B的校正因子为1.11;新霉胺对新霉素B的校正因子为1.36。
因此,在计算有关物质中新霉胺的量时应采用加校正因子的自身对照法。样品中存在的其他杂质,含量较低,对其进行单独制备较为困难,另外从结构上看,基本含有新霉素的3个母环,推测这些杂质响应与新霉素B一致,故采用自身对照法对其他杂质进行计算。

Figure3 Chromatograms of specificity of HPLC-PAD
3.4 检测限与定量限
当新霉素B与新霉素C质量浓度分别为0.07 μg/L时,信噪比均约为3;质量浓度分别为0.14 μg/mL时,信噪比均约为10,按进样量25 μL换算,则新霉素B和新霉素C的检测限均为1.75 ng,定量限均为3.5 ng。
3.5 重复性
取同一批号硫酸新霉素样品6份以及新霉素标准品按照“2.2.4”项含量测定溶液方法配制,按照“2.1”项色谱条件进样分析,新霉素B含量的RSD(n=6)为0.6%,新霉素C含量的RSD(n=6)为0.6%。
取同一批号硫酸新霉素样品6份按照“2.2.5”项有关物质测定溶液方法配制,按照“2.1”项色谱条件进样分析,各杂质的含量的RSD均符合要求,杂质总量RSD(n=6)为1.10%,方法重复性良好。
3.6 耐用性考察
取一批硫酸新霉素样品,按“2.2.5”项下方法配制溶液,略微改变“2.1”项下色谱条件的参数如:柱温、流速,分别进样。各峰分离良好,方法的耐用性良好。
4 方法的应用
4.1 杂质谱研究
采用本研究建立的色谱条件,运用在线膜抑制技术[16],实现了液质联用,对硫酸新霉素中的杂质进行结构推定和来源分析,发现硫酸新霉素原料药中除有新霉胺,还有核糖霉素、巴龙霉素及其异构体、巴龙霉胺、乙酰新霉胺、乙酰新霉素B及其异构体等其他杂质,主要降解杂质为新霉胺[17],推定结果见表1。
Table1 Presumed results of mass spectrometry for impurities of neomycin sulfate

NumberPresumed resultsAImpurity related to neomycinBImpurity related to paromamycinCParomamineDUnknown 1ENeamineFAcetyl-neamineGRibostamycinHImpurity related to ParomamycinIUnknown 2JParomamycin IKParomamycin IILAcetyl-neomycin B isomerMAcetyl-neomycin B
4.2 有关物质测定结果
按“2.2.5”项下方法制备有关物质溶液,按“2.1”项下色谱条件测定,有关物质典型色谱图见图4,结果见表2。有关物质除新霉胺外采用不加校正因子的主成分自身对照法计算。
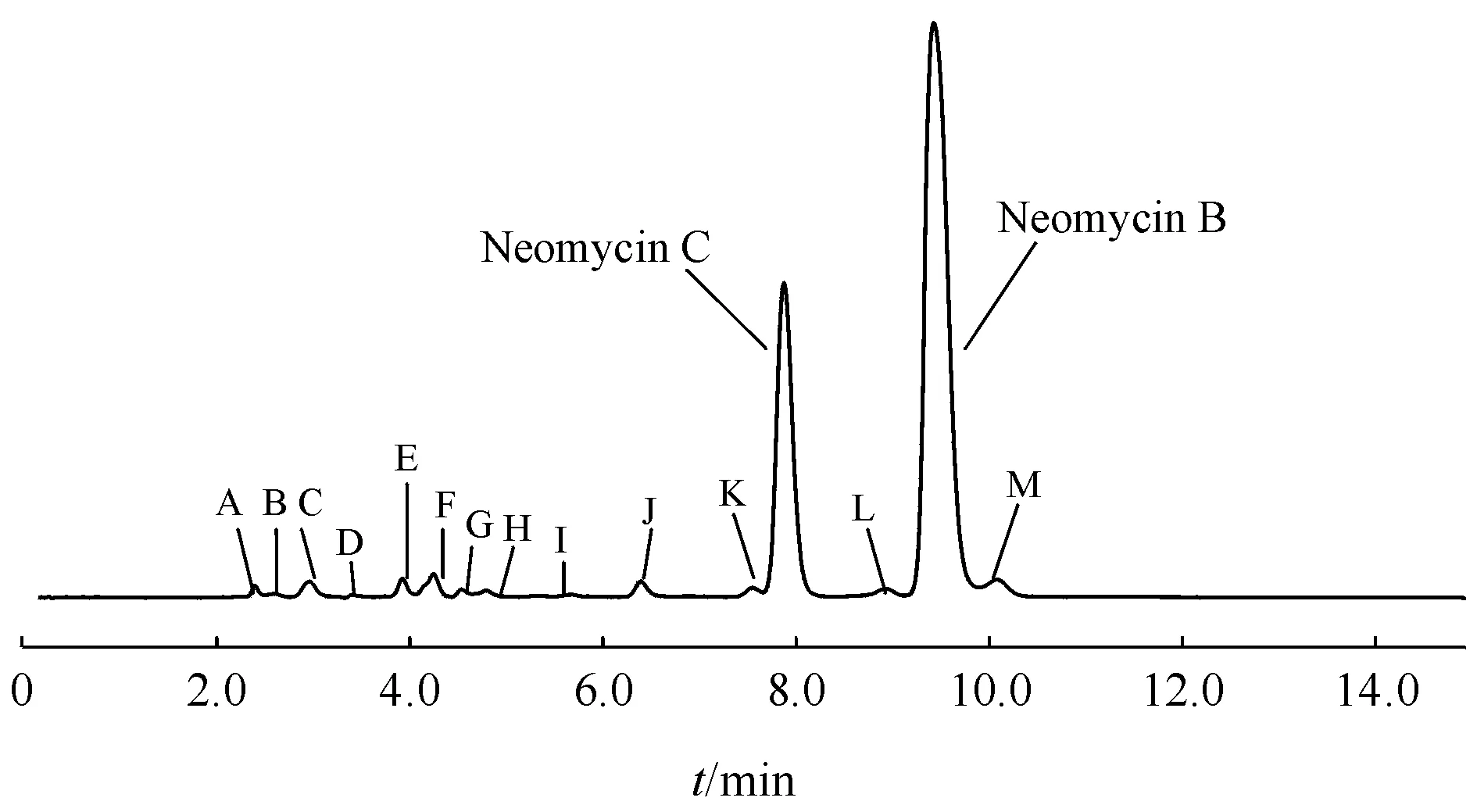
Figure4 Typical chromatogram of related substances for neomycin sulfate
Table2 Result of related substance in 12 batches of raw materials

ManufacturersSamplenumberA/%B/%C/%D/%E/%F/%G/%H/%I/%J/%K/%L/%M/%Total/%Maximum single impurity*/%A10.220.060.680.070.310.710.190.200.210.750.200.480.284.28J(0.75)20.290.110.530.061.830.420.120.090.150.640.110.320.124.79E(1.83)30.170.111.110.141.021.340.240.210.181.020.270.630.556.69F(1.34)40.170.110.270.060.610.370.430.100.160.720.260.320.233.64J(0.72)50.230.100.340.050.340.480.300.130.161.070.650.370.344.48J(1.07)60.160.211.020.101.101.220.180.120.121.440.390.600.436.79J(1.44)B70.060.261.330.190.691.680.130.260.261.981.360.460.478.96J(1.98)80.060.231.390.190.641.930.130.300.171.951.460.480.719.47J(1.95)90.050.171.410.070.671.670.120.350.181.611.170.510.768.58F(1.67)100.030.251.030.140.671.820.100.210.151.050.650.690.887.50F(1.82)110.030.110.930.060.561.280.080.190.220.980.740.580.786.41F(1.28)120.050.150.960.060.751.440.110.200.141.060.750.620.856.94F(1.44)
A:Impurity related to neomycin;B:Impurity related to Paromamycin;C:Paromamine;D:Unknown 1;E:Neamine;F:Acetyl- neamine;G:Ribostamycin;H:Impurity related to Paromamycin;I:Unknown 2;J:Paromamycin I;K:Paromamycin II;L:Acetyl-neomycin B isomer;M:Acetyl-neomycin B
*:Maximum single impurity of sample
Tip:The sample of enterprise B was over effective period for the research
由两个企业各6批共12批原料药的检测结果可见,各批次检出来的杂质种类一致,仅杂质量不同,各批原料药样品的单个杂质量基本都大于0.1%,最大杂质多集中在巴龙霉素Ⅰ;部分批次乙酰新霉胺为最大杂质,其余新霉素相关杂质、巴龙霉素相关杂质和核糖霉素略低。考虑本品为多组分发酵的抗生素,结合欧洲药典杂质限度要求和样品的实际测定情况,将杂质限度定为“新霉胺不得大于2.0%,单个杂质不得大于5.0%,杂质总量不得大于15.0%”。12批样品有关物质结果均符合规定。
4.3 含量测定结果及量效一致性研究
按“2.2.4”项下方法制备含量测定溶液,按“2.1”项下色谱条件,用经标化的新霉素标准品以外标法计算新霉素B和新霉素C的量(以干燥品计),测定结果见表3。
由于收录硫酸新霉素的国内外药典中含量测定方法均采用抗生素微生物检定法,测定结果采用效价单位表示,而仪器法测得的含量为质量单位,两个结果无法进行简单的转换,因此,需通过实验得到量效转换系数,再通过计算将质量含量转换成效价含量,这一研究称为量效一致性研究。参考USP[18-19]相关实验指导原则及文献[20]中关于硫酸庆大霉素的研究思路,采用本研究建立的HPLC-PAD法对12批样品中新霉素B和新霉素C的质量含量进行测定,经新霉素B和新霉素C的量效转换系数计算,转换为效价含量后,与采用微生物鉴定法测定的效价值对比,经统计学分析无显著性差异,表明含量测定结果真实可靠,完成了仪器测定法对微生物效价测定法的替代。
Table3 Result of content of neomycin sulfate in 12 batches of raw materials

BreedManufacturerSampleContent of neomycin C/%(By dry substance)Content of neomycin B/%(By dry substance)Neomycin sulfateA114.949.9215.950.7316.049.4411.654.2511.053.3615.549.2Neomycin sulfateB78.052.387.053.197.851.4109.450.8118.751.4128.851.4
Tip:The sample of enterprise B was over effective period for the research
5 讨 论
5.1 色谱柱的选择
相比于其他氨基糖苷类抗生素,硫酸新霉素的极性较大,在一般的ODS柱上保留较弱,EP9.0中有关物质测定方法流动相为含0.6%氢氧化钠的2%三氟乙酸水溶液,不含有机相,pH约为1.2,对色谱柱的耐水性及耐酸性要求较高。本研究采用EP9.0的方法,考察了3种亲水极性柱:Agilent Zorbax SB-Aq(4.6 mm×250 mm,5 μm),Waters Xselect T3(4.6 mm×250 mm,5 μm),Thermo AcclaimTMAmG C18(4.6 mm×250 mm,3 μm)对硫酸新霉素的分离情况。结果表明,Agilent Zorbax SB-Aq和Waters Xselect T3这两根色谱柱对新霉素B、新霉素C及各杂质间的分离较差,且检出的杂质个数少。Thermo AcclaimTMAmG C18色谱柱为Thermo公司专门针对氨基糖苷类抗生素开发的色谱柱,其对新霉素B、新霉素C及各杂质均能得到较好的分离,主峰峰形对称,检出的杂质个数较多,因此采用该色谱柱来进一步优化硫酸新霉素HPLC-PAD检测方法。
5.2 流动相的优化
在重复EP9.0有关物质测定方法时,发现新霉素B与其前相邻杂质分离不佳(图5)。尝试在流动相中降低三氟乙酸浓度,使新霉素B及其前相邻杂质出峰变慢,以改善二者的分离度,但此时新霉素B又会包裹其后相邻的杂质。同样通过调整氢氧化钠的浓度也未达到满意的结果。参考硫酸依替米星[21-23]的研究思路,尝试采用五氟丙酸替代三氟乙酸,但硫酸新霉素两个主峰保留过强,分析时间太长。进一步在三氟乙酸存在的基础上加入少量五氟丙酸,两种离子对试剂对不同色谱峰保留时间形成协同影响,且因流动相中存在氢氧化钠提高了pH,对峰型及各杂质间的分离均有改善。通过对两种离子对试剂不同配比的考察,最终确定流动相为含0.01%五氟丙酸及0.6%氢氧化钠的2%三氟乙酸溶液,在此条件下,新霉素B与前后相邻杂质(杂质L、杂质M)的分离良好,其他杂质分离度均符合要求,并且比EP9.0方法多分离出杂质K,分离检测能力有明显改善。
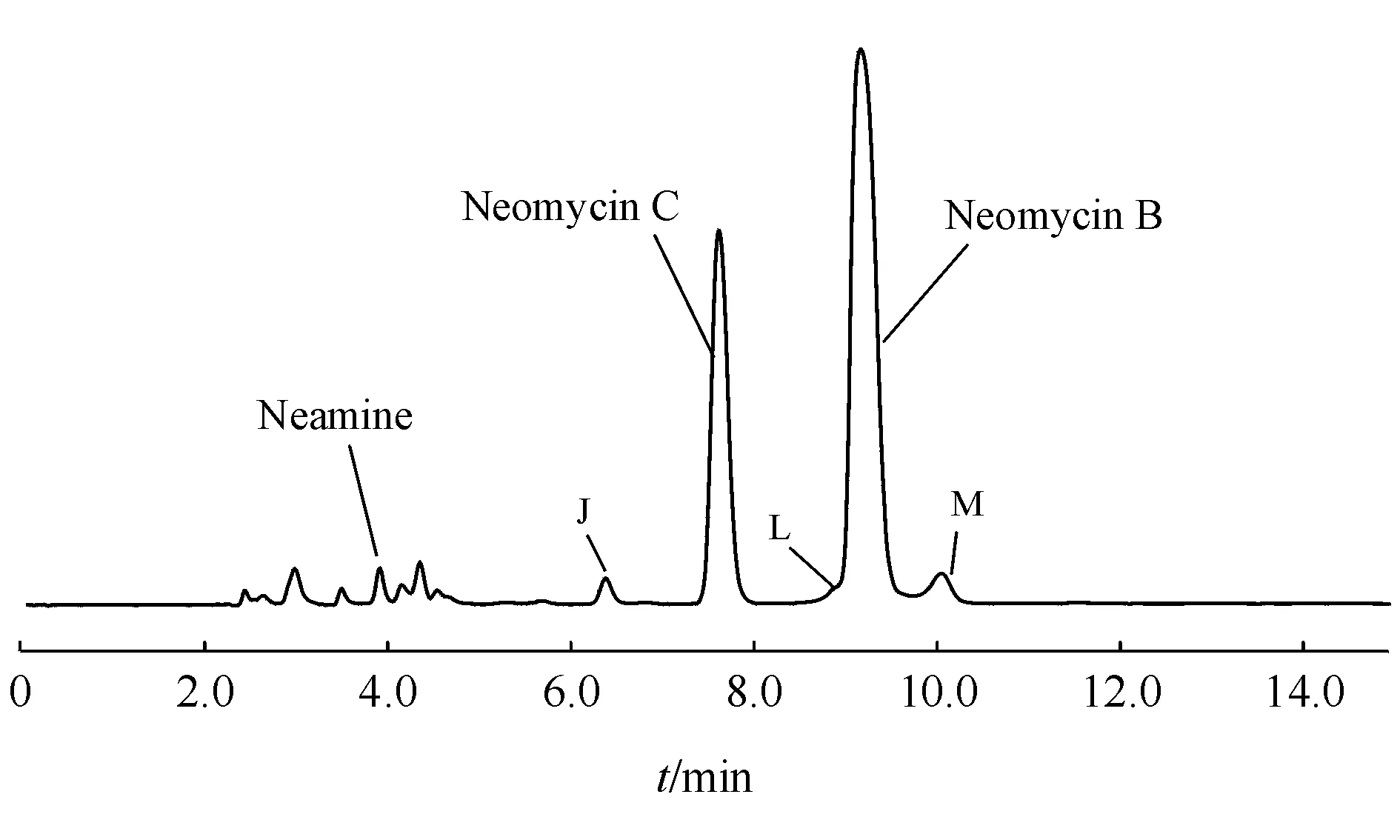
Figure5 Chromatogram of related substances in EP9.0 by Thermo AcclaimTMAmG C18
6 结 论
本次建立的HPLC-PAD方法专属性强、灵敏度高、重复性好,将方法用于实际样品的有关物质及含量测定,方便快捷,结果准确可靠,该方法具有良好的应用前景,可以作为补充中国药典硫酸新霉素标准的推荐方法。
猜你喜欢
承德医学院学报(2022年2期)2022-05-23
硫酸工业(2021年8期)2021-12-26
新疆钢铁(2021年1期)2021-10-14
艺术品鉴(2020年6期)2020-12-06
文萃报·周五版(2020年24期)2020-06-22
理科考试研究·高中(2017年7期)2017-11-04
中学生理科应试(2017年2期)2017-04-01
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
人民周刊(2016年11期)2016-06-30
