
Multisystem smooth muscle dysfunction syndrome in a Chinese girl:A case report and review of the literature
2019-04-25 01:04SaiNanChenYuQingWangChuangLiHaoYanHongLuWuJunJiangChunYanGaoMinWu
World Journal of Clinical Cases 2019年24期
Sai-Nan Chen, Yu-Qing Wang, Chuang-Li Hao, Yan-Hong Lu, Wu-Jun Jiang, Chun-Yan Gao, Min Wu
Sai-Nan Chen, Yu-Qing Wang, Chuang-Li Hao, Yan-Hong Lu, Wu-Jun Jiang, Chun-Yan Gao, Min Wu, Department of Respiratory Medicine, Children’s Hospital of Soochow University, Suzhou 215000, Jiangsu Province, China
Corresponding author: Yu-Qing Wang, MD, Chief Doctor, Department of Respiratory Medicine, Children’s Hospital of Soochow University, No. 303, Jingde Road, Suzhou 215000,Jiangsu Province, China. wang_yu_qing@126.com
Abstract
Key words: Multisystem smooth muscle dysfunction syndrome; Gene mutation;Congenital mydriasis; Patent ductus arteriosus; Case report
INTRODUCTION
Multisystem smooth muscle dysfunction syndrome (MSMDS) is a genetic disease caused mostly by mutation of the actin alpha 2 (ACTA2) gene p.R179H. Its clinical manifestation is characterized by fixed dilated pupils, patent ductus arteriosus (PDA),thoracic aortic aneurysm, pulmonary artery hypertension, cerebrovascular disease,white matter lesions, hypotonic bladder, intestinal malrotation, hypoperistalsis and so on. The ACTA2 gene encodes an isoform of α-actin. Mutations in this gene lead to disruption of smooth muscle cells (SMC)-dependent organs[1,2]. Milewicz et al[1]first reported six patients with a de novo missense mutation in the ACTA2 gene and MSMDS in 2010. In China, the first case of MSMDS was not reported until 2017 by Zhou et al[3]. Since the ACTA2 gene was first identified as the pathogenic gene of MSMDS in 2010, only 32 patients have been reported, according to the databases of PubMed, Wanfang, China National Knowledge Infrastructure, and VIP with the key words “multisystem smooth muscle dysfunction syndrome” and ‘‘ACTA2” from 1980s to 2018. Three different mutation loci have been reported to cause MSMDS, of which Arg179His is the most common mutation. Other rare mutations are Arg179Cys and Arg179Leu[2,4-7]. The Arg179His substitution is associated with the neurovascular phenotype, while the Arg179Cys mutation is especially related to brain development[6]. The most severe form is ACTA2 Arg179His. The discovery of this mutation has been invariably associated with a high risk of infant mortality and poor prognosis[6]. Here, we report one Chinese infant with MSMDS with a heterozygous ACTA2 Arg179His substitution who presented with patent foramen ovale, congenital nonreactive mydriasis, dyspnea, development delay, and abnormal signals in magnetic resonance imaging (MRI) of the brain. In addition, we further review the literature regarding MSMDS patients from the 1980s to 2018s. The clinical features of all identified MSMDS patients are summarized.
CASE PRESENTATION
Chief complaints
A girl aged 9.6 months was admitted to Children’s Hospital Soochow University in May 2018 due to recurrent cough for more than half a month, which was aggravated by shortness of breath and dyspnea for three days.
History of past illness
She had experienced pneumonia at birth and suffered from growth retardation.
Personal and family history
The child was born by cesarean section at 35 + 2 wk. Her father and mother were healthy. Her twin sister died in April 2018 of lung infection after an operation for“atrial septal defect and ventricular septal defect”.
Physical examination
She weighed 9 kg, her height was 65 cm, her body mass index was 21.3, and she presented with shortness of breath and dyspnea. Blue-purple plaques could be seen on her right face, buccal mucosa, oral tongue, and palate. Her bilateral pupils were fixed and dilated with a diameter of 5 mm. Crackles and wheezing rales were present in bilateral lungs. A grade II/6 murmur was audible on the left sternal margin. No clubbed digits were found.
Laboratory examinations
Routine blood examination showed a white blood cell count of 8.17 × 109/L, and the C reactive protein concentration was normal. Nasopharyngeal secretion examination revealed positive respiratory syncytial virus and negative sputum culture.
Imaging examinations
Chest high-resolution computed tomography revealed inhomogeneous lung transparency, obvious exudative lesions, and marked thickening of lung fissures(Figure 1). Cranial magnetic resonance imaging showed abnormal signals in the centrum ovale majus and bilateral periventricular regions (Figure 2). Abdominal ultrasonography suggested gallstones (Figure 3). Echocardiography only showed patent foramen ovale at the beginning of the disease, but the diameter of the pulmonary artery gradually widened during continuous follow-up (Figure 4).Bronchoscopy revealed redness and edema of the bronchial mucous membranes accompanied by phlegm spots and bronchomalacia (Figure 5).
随着《长江经济带发展规划纲要》的实施,沿江省市农业生态环境恶化问题得到初步遏制。同时我们也清醒看到,长江水生生物生存环境日趋恶化,生物多样性指数持续下降,长江江豚、中华鲟等极度濒危,刀鲚、暗纹东方鲀等重要渔业资源面临全面衰退;化肥、农药等农业投入品过量使用,畜禽粪便、农作物秸秆、农田残膜等农业废弃物不合理处置,农业面源污染治理难度大。推进长江经济带农业农村绿色发展,强化水生生物多样性保护,严格控制农业面源污染,实现投入品减量化、生产清洁化、废弃物资源化、产业模式生态化,才能加快补齐农业农村生态环境保护短板。
Gene sequence analysis
The patient underwent genetic analysis and the ACTA2 c.536G>A (p.R179H)heterozygous mutation was found, but her parents’ sample identifications were negative (Figure 6).
FINAL DIAGNOSIS
MSMDS.
TREATMENT
Her symptoms improved after oxygen therapy, cefazoxime administration for 3 wk,and antiasthenic and low-dose corticosteroid treatment for 3 wk.
OUTCOME AND FOLLOW-UP
After being discharged from our hospital, the child was followed monthly in the outpatient clinic. We administered low-dose azithromycin and low-dose corticosteroid anti-inflammatory treatment. We performed regular examinations of respiratory rate, oxygen saturation, and high-resolution computed tomography of the chest to evaluate the pulmonary disease regression/progression, and echocardiography to evaluate the function of the heart. She had pulmonary infection three times, and her latest echocardiographic results suggested pulmonary artery dilatation. She weighed 10 kg, her height was 72 cm, and her body mass index was 19.3. Her overall situation has remained well to date.
DISCUSSION
Tables 1-4 summarizes the systemic features of the reported patients with MSMDS worldwide[8-16]. SMCs are widely distributed in the gastrointestinal tract, urinary tract,respiratory tract, uterus, iris, and other body parts. Their contraction and relaxation are key to the normal operation of blood vessels as well as the digestive system,respiratory system, and urogenital system. MSMDS is mainly caused by p.R179H mutations of the ACTA2 gene in aortic smooth muscle. This disease is a systemic disorder of smooth muscle function and manifests in a variety of ways, including fixed dilated pupils, PDA, thoracic aortic aneurysm, pulmonary artery hypertension,cerebrovascular disease, white matter lesions, hypotonic bladder, intestinal malrotation, and hypoperistalsis. Other rare symptoms, including pulmonary cystic disease, venous thrombosis, cleft palate, hypothyroidism, and deepened skin folds,need further study to confirm the relationship with this mutation.

Figure 1 Chest high-resolution computed tomography images. A chest high-resolution computed tomography scan showed inhomogeneous lung transparency and obvious exudative lesions, and some lung fissures were markedly thickened in the lung of the patient.
In 2010, Milewicz et al[1]first reported a case of MSMDS associated with the p.R179H mutation of the ACTA2 gene. This mutation caused systemic smooth muscle dysfunction, leading to aortic and cerebrovascular diseases, fixed dilated pupils,hypotonic bladder, intestinal malrotation, and pulmonary artery hypertension. In 2017, Zhou et al[3]reported the first case of MSMDS in China. Our case is the second case of MSMDS diagnosed by genetic testing in China. Similar to the reported cases,our patient had congenital mydriasis accompanied by loss of direct light reflex. Iris tissue dissociation was considered an early manifestation of these pupil symptoms[5].Fundus examination showed no retinal vascular tortuosity in our patient. Moller et al[5]reported the ophthalmic characteristics of three patients with MSMDS in detail.They believed that the retinal vascular tortuosity was due to the lack of elastic lamina in the retinal artery wall, and SMC proliferation resulted in vascular obstruction.Furthermore, the incidence of retinal vascular tortuosity tended to increase with age,which may be related to changes in the vascular wall and the loss of contractility. In cardiovascular diseases, our patient had patent foramen ovale and pulmonary artery dilatation without PDA or pulmonary hypertension, which is different from the reported cases. PDA was present in all reported cases. The ductus arteriosus, similar to the muscular artery, is a duct between the aorta and the pulmonary artery. SMCs play a major role in catheter closure, and their dysfunction leads to the persistence of ductus arteriosus. It has been reported that pulmonary hypertension may be due to the combined action of large PDA and pulmonary parenchymal diseases and that the repair of PDA is essential for the recovery of pulmonary parenchymal diseases[15]. In a long-term follow-up study of three MSMDS patients by Yetman et al[13], they found that the aorta dilated progressively with age, which may be related to the change in α protein structure caused by ACTA2 mutation leading to the decrease in aortic contractility. However, the role of drug therapy in preventing these cardiovascular lesions is still unknown. With regard to cerebrovascular diseases, our patient received cranial MRI, which showed abnormal signals in the centrum ovale majus and bilateral periventricular regions. As reported, MSMDS mostly began as a pulmonary disease.Our patient had a history of "neonatal pneumonia" at birth. When she was 9 mo old,she suffered from shortness of breath, wheezing, and dyspnea and needed long-term oxygen therapy. Roulez et al[10]holds that decreased SMC function of pulmonary alveoles leads to tachypnea at birth, pulmonary hypertension, asthma, bronchiectasis,and emphysema. However, the relationship between the ACTA2 gene causing MSMDS and lung pathology has not been well elucidated. Prabhu et al[15]believe that pulmonary vascular changes may be caused by pulmonary hypertension secondary to PDA or by elevated pulmonary pressure caused by structural disorders of abnormal alveolar growth. However, in the case of the absence of intimal hyperplasia, intimal hypertrophy of small muscle arteries may be caused by the hypercontraction of arterial smooth muscle. Our patient had skin and mucosal symptoms, which manifested as cyan-purple plaques on her right face, buccal mucosa, oral tongue, and palate. Richer et al[14]first reported a case of skin abnormalities in which wrinkles of ankles, knees, buttocks, and elbows were markedly deepened. This may be related to the dysfunction of myofibroblasts expressing ACTA2 in the skin.

Figure 2 Cranial magnetic resonance imaging of the patient. Magnetic resonance imaging showed abnormal signals in the centrum ovale majus and bilateral periventricular regions.
Tables 1-4 summarizes the systemic features of the reported patients with MSMDS worldwide. Among the MSMDS patients, the youngest was a neonate, and the oldest was 31 years old. Twenty-three (71.9%) were female, and nine (28.1%) were male.Except for cardiovascular and cerebrovascular diseases, these patients also experienced dysfunction of other SMC-dependent organs, including depressed systolic performance of the bladder and gastrointestinal tract, resulting in hypotonic bladder, hydronephrosis, hypospadias, intestinal malrotation, hypoperistalsis, and gallstones. As shown in Tables 1-4, all patients tested positive for the ACTA2 gene mutation. The R179H variant was the most common mutation type (81.25%), followed by R179C (12.5%) and R179L (6.25%). This mutation has always been reported to be sporadic, since few patients can survive to reproductive age, and none of the parents who had undergone genetic testing were identified as carriers. By the time of submission, five deaths had been reported in all cases. Our patient was given oxygen therapy and low-dose corticosteroid treatment after discharge. She suffered from pulmonary infection three times, and her latest echocardiographic results suggested pulmonary artery dilatation without pulmonary hypertension.
ACTA2, located in the long arm of chromosome 10-10q23.31, is composed of nine exons and codes for actin alpha 2, a major contractile protein in vascular smooth muscle cells. ACTA2 is one of six actin subclasses in mammals, mainly distributed in muscle cells and is the main component of contractors. By the interaction of the contractile element α actin (ACTA2 coding) and β myosin heavy chain (MYH11 coding), SMCs can achieve contraction to regulate blood pressure and blood flow.Therefore, there are many overlapping clinical manifestations of megacystismicrocolon-intestinal hypoperistalsis syndrome (MMIHS) caused by the mutation of MYH11 and MSMDS caused by the mutation of ACTA2. Yetman et al[17]reported a case of MMIHS with congenital mydriasis, PDA, pulmonary hypertension, aortic dilatation, intestinal malrotation, and hypoperistalsis but no cerebrovascular abnormalities. Therefore, for children with clinical characteristics such as congenital mydriasis and PDA, genetic testing should be carried out as soon as possible. At the same time, eye fundus examination, echocardiography, and cranial MIR should also be performed to observe multisystem lesions.
CONCLUSION
In conclusion, MSMDS is a rare genetic variant disease. According to the literature,the hotspot variant is the heterozygous mutation of the ACTA2 gene c.536C>T(p.R179H). Early genetic testing is essential for the optimal management of suspected clinical cases. In addition to early detection of hotspot variation in these patients,clinicians need to examine and evaluate the patients’ cerebrovascular, cardiovascular,and pulmonary complications to make early diagnosis and treatment.

Table 1 Clinical characteristics of multisystem smooth muscle dysfunction syndrome patients worldwide

Table 2 Clinical characteristics of multisystem smooth muscle dysfunction syndrome patients worldwide

Table 3 Clinical characteristics of multisystem smooth muscle dysfunction syndrome patients worldwide

+: Present; -: Absent; blank: Unknown; PDA: Patent ductus arteriosus; ASD: Atrial septal defect; PAH: Pulmonary artery hypertension; TAA: Thoracic aortic aneurysm; AAD: Ascending aorta dilatation; PAD: Pulmonary artery dilatation; WM: White matter; RRTI: Recurrent upper respiratory tract infection; H: R179H; C: R179C; L: R179L; D: Death.

Table 4 Clinical characteristics of multisystem smooth muscle dysfunction syndrome patients worldwide

+: Present; -: Absent; blank: Unknown; PDA: Patent ductus arteriosus; ASD: Atrial septal defect; PAH: Pulmonary artery hypertension; TAA: Thoracic aortic aneurysm; AAD: Ascending aorta dilatation; PAD: Pulmonary artery dilatation; WM: White matter; RRTI: Recurrent upper respiratory tract infection; H: R179H; C: R179C; L: R179L; D: Death.

Figure 3 Abdominal ultrasonography images. Abdominal ultrasonography revealed a strong echo mass of approximately 4 × 6 in size in the gallbladder.

Figure 4 Echocardiography. Echocardiography showed a widening pulmonary artery.

Figure 5 Bronchoscopic findings. Bronchoscopy revealed redness and edema of the bronchial mucous membranes. Phlegm spots were observed in the left lung,and bronchomalacia was observed in bilateral lungs.
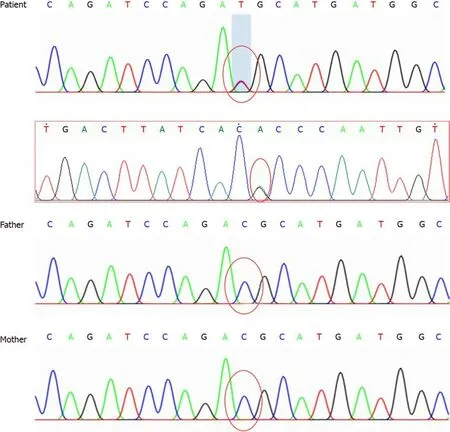
Figure 6 Genomic sequence of the patient. Sequencing map of the actin alpha2 (ACTA2) gene showed a C-T heterozygous variation at nucleotide 536 in the coding region of the ACTA2 gene, causing 179 amino acid changes. No genetic mutations were found in her parents.
猜你喜欢
中国农业科学(2022年17期)2022-09-19
商品与质量(2021年43期)2022-01-18
当代水产(2021年10期)2022-01-12
云南农业科技(2021年6期)2021-12-30
中国农资(2021年11期)2021-12-14
今日农业(2021年1期)2021-11-26
当代水产(2021年2期)2021-03-29
农业工程(2020年9期)2020-12-13
短篇小说(原创版)(2017年6期)2017-08-29
小小说月刊(2017年8期)2017-08-14
 World Journal of Clinical Cases2019年24期
World Journal of Clinical Cases2019年24期
- World Journal of Clinical Cases的其它文章
- Polyunsaturated fatty acids and DNA methylation in colorectal cancer
- lmpact of resection margins on long-term survival after pancreaticoduodenectomy for pancreatic head carcinoma
- Arthroscopy combined with unicondylar knee arthroplasty for treatment of isolated unicompartmental knee arthritis: A long-term comparison
- lntact, pie-crusting and repairing the posterior cruciate ligament in posterior cruciate ligament-retaining total knee arthroplasty: A 5-year follow-up
- Community-acquired pneumonia complicated by rhabdomyolysis: A clinical analysis of 11 cases
- Dissection and ligation of the lateral circumflex femoral artery is not necessary when using the direct anterior approach for total hip arthroplasty