
磷掺杂石墨相氮化碳的制备及对磺胺噻唑的可见光催化性能研究
2019-04-09 09:05:00丁任丽郑诗瑶
生态与农村环境学报 2019年3期
唐 荣,丁任丽,郑诗瑶
(1.江苏开放大学环境与生态学院,江苏 南京 210036; 2.南京理工大学环境与生物工程学院,江苏 南京 210094)
随着医药水平的不断提高,抗生素在医药中的应用愈来愈普遍。磺胺噻唑(sulfathiazole,ST)是一种典型的磺胺类抗生素,被广泛应用于临床医学、畜牧和水产等行业[1]。由于磺胺噻唑在生物体内较难代谢,会通过生物排泄等途径进入环境,造成环境污染;此外,磺胺噻唑易被农作物吸收,通过食物链、食物网等途径进入人体,累积到一定量后,会危害人体的免疫系统,影响造血功能[2]。传统的水处理方法较难完全去除水体中的磺胺噻唑,关于残余磺胺类抗生素无害化处理的研究已引起广泛关注。
自1972年FUJISHIMA等[3]通过紫外光照射TiO2单晶电极获得H2和O2并发现了光电催化分解现象以来,光催化技术一直备受关注。光催化技术可实现太阳能的高效转化和储存,在一定条件下可驱动重要的化学反应,在解决能源环境问题上有着较大的潜力[4]。大量研究结果表明,染料、有机卤化物、农药、油类和氰化物等污染物都能通过光催化反应得到有效处理,进一步矿化为无机小分子物质,从而减小或消除其对环境的污染[5]。然而,传统的半导体光催化剂存在光谱响应范围窄、光催化量子效率低、载流子迁移率小和稳定性差等缺点[6]。因此,探索性能优异、价格低廉、结构稳定的可见光催化剂,对解决环境问题具有重要意义。
石墨相氮化碳(graphitic carbon nitride,简称g-C3N4)是近年来研究较多的一种新型非金属半导体可见光光催化剂,仅含C、N两种元素。g-C3N4合成成本比贵金属催化剂低廉,独特的聚合特性又使其能带在原子水平上可调;同时,g-C3N4的纳米结构可塑性强,有利于其与其他半导体间形成异质结构[7-8]。然而,进一步研究发现,g-C3N4存在可见光利用效率较低、光生载流子易复合、光催化量子效率低等缺点,限制了其在环境治理中的应用[9]。近年来,针对g-C3N4存在的问题,科学家们探索出一系列方法调控其表面形貌、能带结构和电子结构,从而提高其光催化性能。这些调控方法主要包括元素掺杂、半导体复合、形貌调控和光敏化等[10]。其中,元素掺杂在提高g-C3N4的光催化性能方面有着巨大优势。元素掺杂主要包括金属掺杂和非金属掺杂。非金属元素掺杂g-C3N4可使催化剂的能带位置得以改变,禁带宽度减小,光谱吸收范围拓宽,催化剂的光催化性能也相应得到改善。目前常用来改性的非金属元素主要有S[11]、O[12]、B[13]、F[14]等。徐赞等[15]提出利用磷酸氢二铵作为磷的前驱体进行掺杂时,P可掺杂至g-C3N4模型中。研究表明,P原子会优先取代g-C3N4中的C原子,从而导致g-C3N4导带升高,表面呈富电子状态,这些电子会与氧气结合形成超氧自由基,使得材料的氧化性增强。利用光催化方法处理染料废水的结果显示,P掺杂的g-C3N4材料对罗丹明B(RhB)的降解效率最高可达97%,是体相g-C3N4降解效率的1.52倍。FENG等[16]通过固体反应路线合成了P掺杂的石墨相氮化碳(P-g-C3N4),该复合材料能够快速降解RhB,同时其产氢速率达到941.80 μmol·h-1·g-1,但该材料的合成条件比较苛刻。QIU等[17]以黑磷作为修饰剂,对g-C3N4进行改性,并采用超声法制备黑磷掺杂的氮化碳薄片(BPCNS)。结果表明,与纯氮化碳薄片(CNS)相比,BPCNS的固氮效果提高5.6倍,但黑磷的稳定性一直是个备受争议的问题。由此可见,对g-C3N4进行P掺杂改性是提高g-C3N4可见光催化活性的有效途径,但如何通过改进方法来实现可控的P掺杂,在提高光催化活性的同时保持材料的稳定性,仍是有待解决的难题。
笔者以一种生物原料——甘油磷脂酰胆碱作为磷的前驱体,经与三聚氰胺混合,通过水热和高温煅烧联合方法制备磷掺杂CN(PCN)。在可见光照射条件下,以抗生素ST为模拟污染物,研究不同掺磷量PCN催化剂可见光催化降解ST的效果,并通过捕获实验进一步探讨PCN光催化降解ST的反应机制。
1 材料与方法
1.1 PCN的制备
1.1.1CN的制备
将10 g三聚氰胺溶解在60 mL去离子水中,搅拌均匀后转移至100 mL反应釜内,于180 ℃条件下水热反应24 h,将水热反应后的材料水洗,烘干;然后将得到的材料置于马弗炉中在500 ℃条件下煅烧2 h之后再在550 ℃条件下煅烧4 h(升温速率为5 ℃·min-1),水洗,烘干,得到粉末状材料,记为石墨相氮化碳(CN)。
1.1.2PCN的制备
将10 g三聚氰胺和Xg(X分别为2、4、6、8)甘油磷脂酰胆碱溶解在60 mL去离子水中,搅拌均匀后转移至反应釜内,于180 ℃条件下水热反应24 h,将水热反应后的材料水洗,烘干;然后将得到的材料置于马弗炉中在500 ℃条件下煅烧2 h之后再在550 ℃条件下煅烧4 h(升温速率为5 ℃·min-1),水洗、烘干,得到粉末状材料记为ω% PCN(ω=X/10)。
1.2 催化剂的表征
X射线衍射(XRD)测定采用德国Bruker公司生产的D8 Advanve型X射线衍射仪,可用于精确地测定物质的晶体结构;傅里叶变换红外光谱(FTIR)测定采用美国赛默飞世尔公司生产的Nicolet iS10傅里叶变换红外光谱仪,用于粉末样品的定性、定量分析及反应研究;采用JEOL JEM 2100型透射电子显微镜(TEM)观察材料的表面形貌和结构特征;紫外可见吸收光谱(UV-vis DRS)测定采用美国赛默飞世尔公司生产的EV 220型紫外-可见分光光谱仪,扫描波长范围为200~800 nm;光致发光光谱(PL)测定采用法国Horiba Jobin Yvon公司生产的FL3-TCSPC型荧光光谱仪,可对样品进行荧光发射光谱和寿命的测试;在Micrometrics ASPS 2020型吸附仪上采用N2吸附脱附方法测定样品的比表面积和孔结构;X射线光电子能谱(XPS)和价带X射线光电子能谱(VBXPS)测定采用美国Thermo ESCALAB 250型X光电子能谱仪,光源为单色Al的Kα射线,功率为150 W,VBXPS用于分析材料的价带电势;莫特肖特基(Mott-Schottky)测定用于分析材料的平带电势,判断导带电势的变化,测试在上海辰华仪器公司CHl660B的电化学工作站上进行,测量条件为以500 W短弧氙灯为光源,以涂有催化剂的导电玻璃(ITO)为工作电极,以Pt电极为对电极,以Ag/AgCl为参比电极,以0.5 mol·L-1Na2SO4水溶液为电解液。
1.3 催化反应活性研究
采用XPA-7型光化学反应仪进行光催化性能测试,以抗生素ST为目标污染物,以500 W氙灯为可见光光源,反应温度为25 ℃,催化剂用量为0.02 g,ST溶液体积为50 mL,质量浓度为30 mg·L-1。具体实验步骤如下:将0.02 g催化剂加入50 mL的ST溶液中,超声使其充分混合,暗吸附30 min后,开启500 W氙灯,稳定后开始计时,按一定时间间隔(30 min)从取样口取样,过滤后测定滤液中ST浓度。采用高效液相法(HPLC)测定滤液中ST浓度,流动相为φ=30%的乙腈水溶液,测定波长为270 nm,柱温为30 ℃。
2 结果与讨论
2.1 催化剂表征
CN和ω% PCN的TEM谱图见图1a~b。由图1a~b可知,CN呈较厚的堆叠状,而60% PCN形貌发生明显变化,呈破碎的薄片状。这是由于在材料制备过程中P的掺杂影响了三聚氰胺的高温聚合反应,使得CN被剥离成薄片,薄片形貌可为光催化提供更多的反应活性位点。CN和ω% PCN的XRD谱图见图1c,CN在衍射角2θ为13.3°和27.6°处出现衍射峰。27.6°处最强峰为特征层间芳香族化合物的堆积峰,对应于CN(002)晶面[18]。小角13.3°处出现微弱峰,对应于CN(100)晶面,是由三嗪结构单元形成的[19]。与CN谱图相比,ω% PCN谱图出峰位置和强度并未发生明显变化,仍保留CN(100)和CN(002)晶面,这表明掺P过程并未改变CN的晶相组成。

CN和ω% PCN的PL谱图见图1e。与CN相比,60% PCN的光谱强度明显降低,表明P原子的引入有效地抑制了光生电子与空穴的复合。这一结果与QIU等[17]报道的结果一致。值得注意的是,80% PCN的光谱强度反而高于CN,这可能是由于P的过量掺杂使得材料处于富电子状态,从而导致光生电子空穴复合率增加。
CN和ω% PCN的UV-Vis DRS谱图见图1f。由图1f可知,CN吸收阈值为494 nm。随着掺P量的增加,20% PCN、40% PCN和60% PCN吸收阈值分别为501、503和510 nm。然而80% PCN的吸收阈值为477 nm,发生了明显蓝移,这可能是由于80% PCN的片层过薄,发生了量子限域效应所致[22-23]。经Kubelka-Munk方法计算,CN和60% PCN的禁带宽度分别为2.50和2.47 eV。由此可见,在一定范围内,P的掺杂有利于缩小催化剂的禁带宽度,拓宽催化剂对光谱的响应范围,从而进一步增强催化剂的光吸收能力。
CN和60% PCN的Mott-Schottky谱图见图2。由图2可知,与CN相比,60% PCN的导带位置发生了明显下移,其中vs. NHE指相对于标准氢电极而言。结合UV-Vis DRS的分析结果可知,60% PCN的价带位置也随之下移。价带位置的下移有利于提高材料的氧化性能,从而促进光催化氧化处理污染物。
由N2吸附脱附表征得到的体相CN比表面积为70 m2·g-1,孔容为0.476 cm3·g-1,而60% PCN比表面积为180 m2·g-1,孔容为0.871 cm3·g-1。可见,60% PCN催化剂的比表面积和孔容较CN均有所上升,表明P的引入为光催化反应提供了更多的活性位点,这与TEM的表征结果相一致。
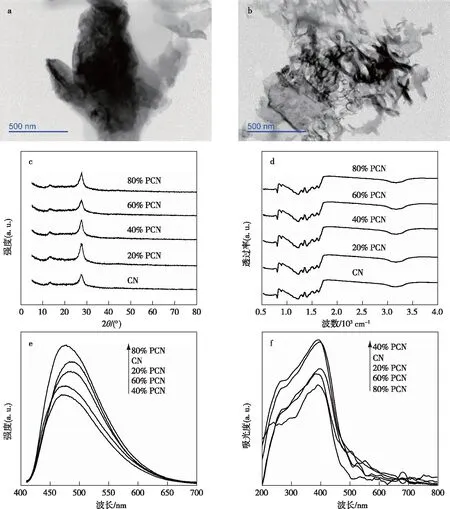
a 体相CN的TEM谱图; b 60% PCN的TEM谱图; c XRD谱图; d FTIR谱图; e PL谱图; f UV-vis谱图。
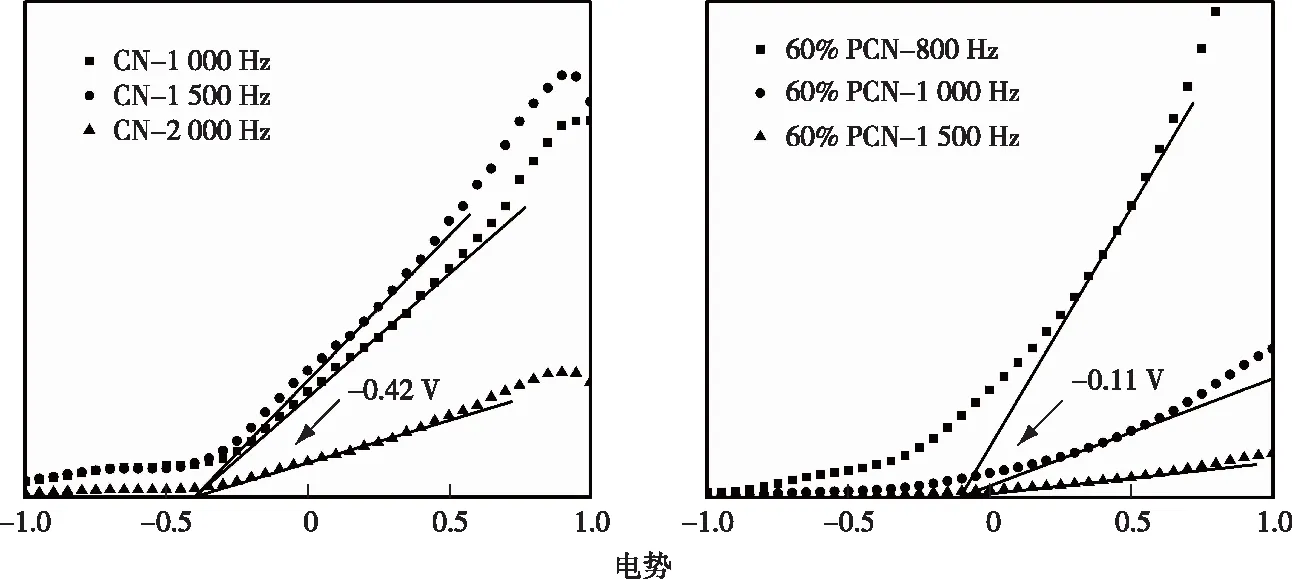
电势单位为V vs. NHE,vs. NHE指相对于标准氢电极而言。
图3 体相CN和60% PCN的XPS谱图
CN和60% PCN的VBXPS谱图和反应机制见图4。利用VBXPS(图4a)计算出的CN和60% PCN的价带(VB)位置分别为1.81和2.13 V,60% PCN的VB位置比CN下移0.32 V。VB位置是决定光催化剂氧化性能的重要参数,一般来说,VB位置越低,材料的氧化性能越强。再根据催化剂的禁带宽度,可以推算出CN和60% PCN的导带(CB)位置分别为-0.69 V和-0.34 V,从而得到CN和60% PCN的能带结构,如图4b所示。深入分析光催化降解ST的反应机制,具体为CN的CB位置为-0.69 V,高于O2/O2·-的电势(-0.046 V vs. NHE),所以能够将材料表面吸附的O2还原成O2·-。但是CN的VB位置为1.81 V,高于·OH/OH-的电势(1.99 V vs. NHE),所以CN价带上的空穴无法将OH-氧化成·OH。同样地,60% PCN的CB位置为-0.34 V,高于O2/O2·-的电势(-0.046 V vs. NHE),所以能够将材料表面吸附的O2还原成O2·-。同时,60% PCN的VB位置为2.13 V,低于·OH/OH-的电势(1.99 V vs. NHE),所以60% PCN价带上的空穴可将OH-氧化成·OH。上述分析结果表明,CN与60% PCN均可产生O2·-,所以在两者光催化降解ST的反应体系中O2·-起氧化作用的现象几乎是一致的;值得注意的是,60% PCN可以将OH-氧化成·OH,而CN不具备氧化产生·OH的能力,所以·OH在光催化反应过程中占主导地位。
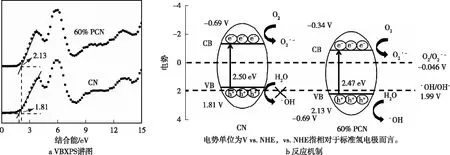
图4 体相CN和60% PCN的VBXPS谱图和反应机制
2.2 催化活性评价
2.2.1可见光催化降解ST
为评估所得催化剂的可见光催化活性,将CN和ω% PCN用于光催化降解ST。CN和ω% PCN在不加滤光片条件下的光催化降解ST的效果见图5。由图5可知,CN和ω% PCN催化剂对ST几乎没有吸附效果;就反应效率而言,60% PCN在不加滤光片条件下的光催化降解ST的效率远优于体相CN。光照2 h后,60% PCN降解ST的效果达到96.4%,而体相CN的降解效果仅为46.2%,这主要归结于:(1)P的掺杂在一定程度上抑制了光生电子空穴的复合,提高了载流子的迁移率;(2)剥离的薄片提高了催化剂比表面积,为光催化提供了更多的反应活性位点;(3)P的掺杂降低了催化剂的价带位置,提高了材料的光催化氧化性能。为了进一步验证该催化剂的可见光催化反应活性,探讨了加滤光片条件下(λ>420 nm)PCN的光催化效果。如图5所示,光照3 h后,体相CN对ST几乎没有降解效果,而60% PCN的降解效果达到42%。该结果进一步证明60% PCN在可见光下具有较好的催化活性。
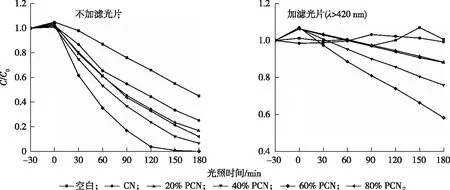
C/C0为随光照时间变化的磺胺噻唑浓度与磺胺噻唑初始浓度的比值,可反映污染物降解效率; 0时刻指30 min暗吸附结束后光照初始时间。
2.2.2光催化稳定性
为了检测催化剂的稳定性能,以60% PCN样品为催化剂,进行5次回收实验。具体过程:在第1次回收实验中,将0.02 g催化剂加到质量浓度为30 mg·L-1的ST溶液(体积为50 mL)中,超声使其充分混合,暗吸附30 min后取样,开启500 W氙灯,稳定后开始计时,光照2 h后从取样口取样,过滤后测定滤液中ST浓度。通过离心方式对样品进行洗涤、回收。在回用5次之后,光催化降解ST的效率依然维持在95%以上,说明60% PCN具有较高的光催化活性稳定性。
2.2.3光催化反应机制
在光催化反应中,半导体光催化剂的氧化性能主要取决于光生空穴(h+)、羟基自由基(·OH)和超氧自由基(O2·-)这3种自由基。为了进一步探究光催化的反应机制,分别以叔丁醇(t-BuOH)、碘化钾(KI)和2,2,6,6-四甲基哌啶醇氮氧自由基(TEMPOL)作为·OH、h+和O2·-的捕获剂,研究60% PCN可见光催化降解ST的主要活性物质。如图6所示,加入TEMPOL后,光催化降解ST的效果与不加捕获剂的效果几乎一致,说明光催化反应体系中起到氧化作用的并非O2·-;加入KI后,光催化降解ST的速率有所减缓,说明h+在光催化反应体系中起到一定的促进作用;加入t-BuOH后,光催化降解ST的效果明显降低,光照3 h后,仅为65%左右,这说明·OH在光催化反应过程中占主导地位。
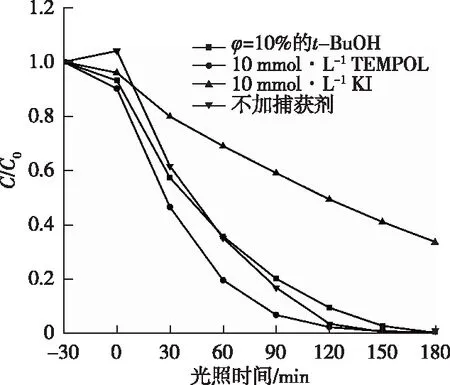
C/C0为随光照时间变化的磺胺噻唑浓度与磺胺噻唑初始浓度的比值,可反映污染物降解效率; 0时刻指30 min暗吸附结束后光照初始时间。
3 结论
以一种生物原料——甘油磷脂酰胆碱作为磷的前驱体,经与三聚氰胺混合,通过水热法和煅烧法联合处理方式制得PCN。与体相CN相比,PCN形貌发生了明显改变,载流子迁移率有所提高,在用于可见光处理抗生素ST时表现出优异性能,所得主要结论如下:
(1)相较于CN,60% PCN对光谱吸收域有所拓宽,且层状结构明显变薄,具有更大的比表面积,同时能很好地抑制光生电子空穴的复合,大大提高催化剂的光催化性能。
(2)改性的60% PCN价带位置发生明显下移,提高了材料的光催化氧化性能。
(3)活性物质捕获实验结果表明,·OH在可见光催化降解ST的过程中占主导地位。
猜你喜欢
家庭影院技术(2019年8期)2019-08-27 02:44:56
石油石化绿色低碳(2019年6期)2019-02-13 09:39:01
陶瓷学报(2019年5期)2019-01-12 09:17:34
三峡大学学报(自然科学版)(2017年1期)2017-03-20 15:30:23
浙江大学学报(工学版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中国资源综合利用(2016年9期)2016-01-22 08:35:22
中国资源综合利用(2016年4期)2016-01-22 08:27:23
燕山大学学报(2015年4期)2015-12-25 02:19:45
中国塑料(2015年4期)2015-10-14 01:09:28
