
高效液相色谱法测定铋镁碳酸氢钠片中甘草酸含量
2019-03-21 01:35:26李志远左文松代明晶
中国药业 2019年6期
李志远,左文松,代明晶
(1.云南省食品药品监督检验研究院,云南 昆明 650106; 2.云南省文山壮族苗族自治州丘北县人民医院,云南 文山 663000)
复方制剂铋镁碳酸氢钠片[1]组方中,甘草流浸膏占比为每1 000片160 g,而现行质量标准中仅收载该成分的鉴别项作为质量控制指标,仅能判断处方中是否含有甘草流浸膏,无法判断厂家在该药品生产过程中是否严格按处方量投料生产;现行标准不能很好地区分不同厂家生产的药品的内在质量。本试验中选择处方中甘草浸膏的有效成分甘草酸为指标成分[2-5],建立测定其含量的高效液相色谱(HPLC)法[6],为该药的质量控制提供定量评价方法。现报道如下。
1 仪器与试药
1.1 仪器
LC-20A型高效液相色谱仪,包括DGU-20A型脱气机、LC-20AT型溶剂输送泵、SIL-20A型自动进样器、CTO-20A型柱温箱、SPD-20A型紫外-可见检测器,LCSolution化学工作站(日本岛津公司);FUNGILAB型超声仪(FUNGILAB仪器公司,功率为180 W,频率为 50/60 kHz);CPA224S/CPA225D型电子天平(赛多利斯科学仪器有限公司)。
1.2 试药
铋镁碳酸氢钠片(昆明积大制药有限公司,批号分别为130301,130305);甘草酸铵对照品(中国食品药品检定研究院,批号为110731);乙腈(赛默飞世尔科技有限公司)、甲醇(默克公司)为色谱纯,水为纯化水,其他试剂均为国产分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:Kromasil C18柱(150 mm ×4.6 mm,5 μm);流动相:乙腈(A)-0.05%磷酸溶液(B),梯度洗脱(洗脱程序见表1);检测波长:237 nm;柱温:30℃;流速:1.0 mL/min;进样量:20 μL。

表1 流动相梯度洗脱表(%)
2.2 溶液制备
取甘草酸铵对照品10.00 mg,精密称定,置25 mL容量瓶中,加50%甲醇溶解并定容,摇匀,作为对照品贮备液。取样品适量,研细,称取0.3 g,精密称定,置50 mL容量瓶中,加50%甲醇适量,超声提取10 min,冷却,加50%甲醇定容,摇匀,经0.45 μm微孔滤膜滤过,取续滤液,作为供试品溶液。按铋镁碳酸氢钠片的处方和工艺制备不含甘草的阴性样品,同法制备阴性对照品溶液。
2.3 方法学考察
系统适用性试验:取2.2项下3种溶液,按2.1项下色谱条件进样测定,记录色谱图(图1)。可见,待测峰与其他峰分离良好,阴性对照品溶液无干扰,理论板数按甘草酸峰计应不低于5 000。
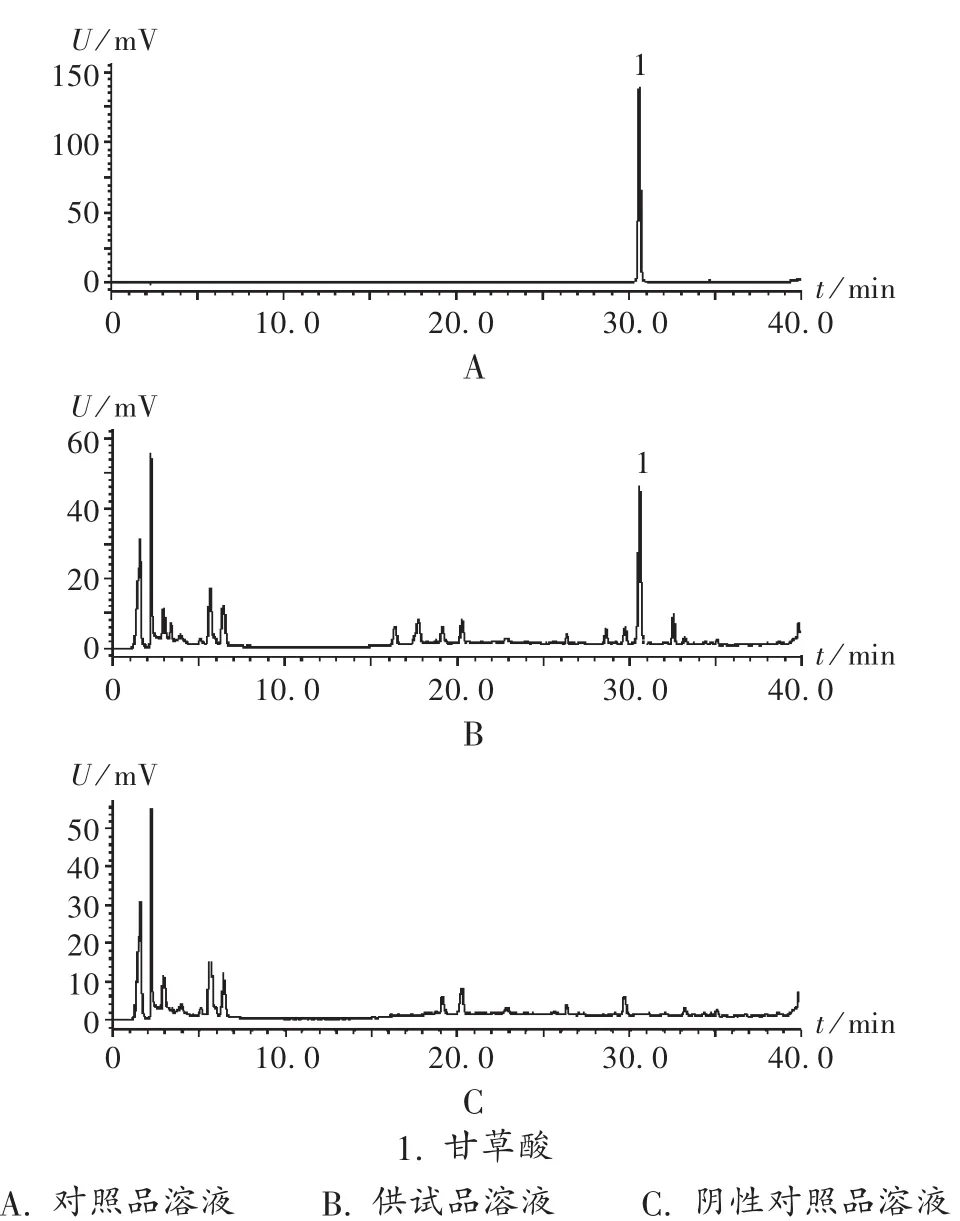
图1 高效液相色谱图
线性关系考察:精密吸取对照品贮备液0.5,1.0,2.0,3.0,5.0 mL,分别置 10 mL 容量瓶中,用 50%甲醇定容至刻度,摇匀,精密吸取20 μL,按2.1项下色谱条件分别进样测定,计算峰面积,记录色谱图,并以甘草酸质量浓度(μg/mL)为横坐标(X)、峰面积(Y)为纵坐标进行线性回归,得回归方程Y=10 498X+4 625.6,R2=0.999 9(n=5)。结果表明,甘草酸质量浓度在20.06~200.60 μg/mL范围内与峰面积线性关系良好。
精密度试验:精密量取同一供试品溶液20 μL,按2.1项下色谱条件连续进样6次,测定峰面积。结果的RSD=0.45%(n=6),表明方法精密度良好。
稳定性试验:取样品(批号为130305)适量,依法制备供试品溶液,精密量取20 μL,分别于室温下放置0,2,4,6,8,12,24 h 时进样测定,测定峰面积。结果的RSD为2.00%(n=7),表明室温下供试品溶液在24 h内稳定。
加样回收试验:取样品(批号为130305)适量,每份0.15 g,共6份,精密称定,置50 mL容量瓶中,加50%甲醇适量,超声提取10 min,分别精密加入对照品溶液,加50%甲醇定容,摇匀,滤过,取续滤液20 μL,按2.1项下色谱条件进样测定,计算回收率。结果见表2。
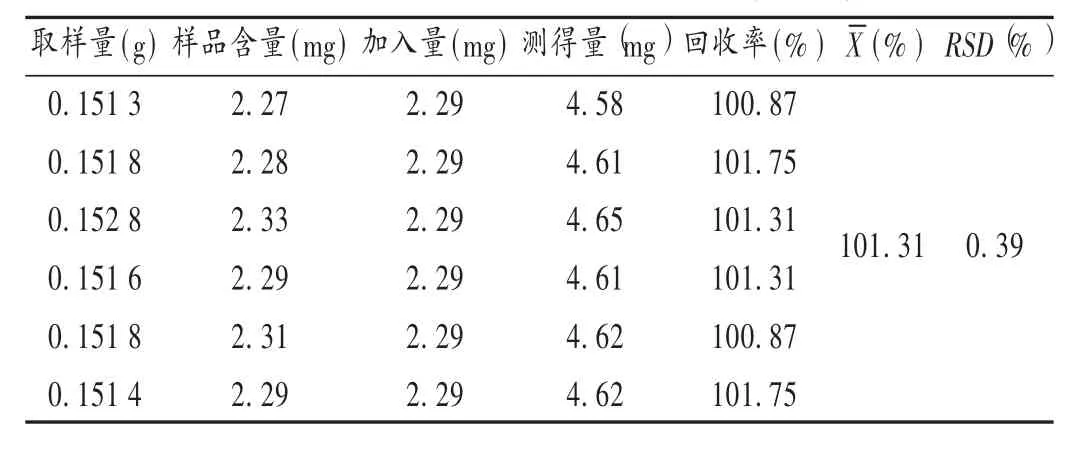
表2 甘草酸加样回收试验结果(n=6)
重复性试验:取样品(批号130301)适量,共6份,同法制备供试品溶液,按2.1项下色谱条件进样测定,记录峰面积。结果的RSD为1.56%(n=6),表明方法重复性良好。
耐用性试验:取供试品溶液,其他色谱条件不变,改变流速、改变柱温、改变色谱柱品牌、相同品牌不同长度,按拟订色谱条件分别进样分析,记录色谱图峰面积。结果的RSD为1.09%(n=5),表明方法耐用性好。
2.4 样品含量测定
取2批样品各适量,研细,称取0.15 g,精密称定,置50 mL容量瓶中,加50%甲醇适量,超声提取10 min,冷却,加50%甲醇定容,摇匀,滤过,取续滤液,按2.1项下色谱条件进样测定,记录色谱图,用标准曲线法以峰面积计算样品中甘草酸含量。结果见表3。

表3 样品含量测定结果(n=7)
3 讨论
3.1 前处理条件优化
提取溶剂确定:取样品0.3 g,精密称定,置50 mL容量瓶中,分别以30%,50%,70%,100%的甲醇溶液作为提取溶剂,按2.1项下色谱条件进样10 μL测定,记录峰面积。结果显示,以50%甲醇溶液作为提取溶剂效果较好。
提取容积确定:称取样品0.3 g,精密称定,分别置25,50,100 mL容量瓶中,以50%甲醇作为提取溶剂,制成不同质量浓度的供试品溶液,按2.1项下色谱条件进样测定,进样量分别为 5,10,20 μL,测定峰面积。结果显示,提取容积为50 mL时效果较好,故选择此法作为样品的提取方法,并超声10 min。
3.2 色谱条件选择
参考2010年版《中国药典(一部)》甘草含量的测定方法,对该流动相体系、检测波长和供试品溶液的制备方法进行了考察[4],结果显示,乙腈-0.05%磷酸溶液流动相体系梯度洗脱所得到色谱图基线较平稳,且分离效果好;甘草酸在237 nm波长处有最大吸收。
3.3 供试品溶液处理方法选择
首先,分别采用30%,50%,70%,100%甲醇溶液进行提取试验,结果发现,50%甲醇提取最完全,故以其作为提取溶剂[5];其次,以不同提取容积对样品进行提取试验,发现以50 mL为提取容积进行提取效果最好。同时选用超声提取方法(50 kHz,180 W,超声时间为10 min)作为供试品溶液的处理方法。
3.4 溶液浓度与对照品选择
本试验中需要配制对照品贮备液,原因在于其浓度较高,且不易改变,而稀释易受环境影响,浓度易变,同时做加样回收试验时每份样品中需加入等量对照品。因此,采用等体积取同一浓度的对照品贮备液的方法可减小其误差[7-8]。本试验中选择甘草酸铵为对照品,在方法学数据采集时均已换算成甘草酸。
综上所述,该方法简便、准确、可靠,可用于铋镁碳酸氢钠片中甘草酸的含量测定。
猜你喜欢
中学生数理化·自主招生(2022年4期)2022-05-09 22:00:23
中国外汇(2019年10期)2019-08-27 01:58:22
中国外汇(2019年10期)2019-08-27 01:58:22
中学化学(2019年4期)2019-08-06 13:59:37
中国外汇(2019年22期)2019-05-21 03:15:02
中国外汇(2019年21期)2019-05-21 03:04:22
Cancer Biology & Medicine(2016年4期)2017-01-13 01:54:45
中国卫生标准管理(2015年4期)2016-01-14 05:16:45
西南医科大学学报(2016年4期)2016-01-03 01:26:29
中国当代医药(2015年33期)2015-03-01 02:09:17
