
戈谢氏病脾切除术1 例报告
2019-03-15 05:55:58陈凌张恒赵礼金
中国普通外科杂志 2019年2期
陈凌,张恒,赵礼金
(遵义医学院附属医院 肝胆外科,贵州 遵义 563000)
患者女,34 岁。因腹痛、腹胀3 d 于2018年5月30日入院。3 d 前患者无明显诱因出现上腹部胀痛,呈持续性疼痛,无放射痛,伴有鼻腔出血,全身皮肤、巩膜轻度黄染。既往史:患者患有“癫痫病”5年,长期口服“丙戊酸钠、卡马西平”治疗,症状可控制。患者于2005年产检时发现“脾大”,未做正规治疗;2006年产女至今就诊于国内多家医院,5年前在北京一家医院曾行骨髓穿刺,并找到戈谢氏细胞,诊断为“戈谢氏病”;近年来肿块逐渐增大。体检:体温36 ℃,心率104 次/min,呼吸22 次/min,血压104/44 mmHg(1 mmHg=0.133 kPa);发育正常,营养较差,急性痛苦面容,神志清晰,全身皮肤轻度黄染,无淤斑、淤点,心肺体检未见异常。腹膨隆,无腹壁静脉曲张,全腹压痛,无反跳痛及肌紧张,触及巨大脾,达中线右侧4 横指,下缘不可及肝未触及,移动性浊音阴性。双下肢轻度凹陷性水肿。神经系统检查无异常。全腹CT 平扫+增强:脾脏明显增大并多发低密度病变及少许高密度影,门静脉增宽,约17 mm。血常规:白细胞2.40×109/L,红细胞2.58×1012/L,血红蛋白59 g/L,血小板35×109/L。
入院后多次予以输血及血液制品(红细胞、血小板、人血白蛋白)对症治疗,并请血液科、营养科会诊,会诊意见:改善患者营养状态后行脾切除术,以纠正血细胞减少,改善巨脾引起的压迫,预防脾破裂、脾梗塞。患者于2018年6年8月行开腹巨脾切除术,术中见:腹腔内少量腹水,脾脏明显增大,充满腹腔,上达隔顶,下至盆腔,色泽暗红,质地较韧,肝左叶明显萎缩。切除脾脏离体称重约9 kg(图1)。术后病理:脾脏脂质贮积病,结合临床病史符合戈谢氏病脾脏改变(图2A-C)。免疫组化结果:CD68(+++);CD20(B 淋巴细胞+);CD3(T 淋巴细胞+);CK(-);Ki-67(5%+);LCA(淋巴细胞+)。特染:脂肪染色(个别细胞+);D-PSA(个别+);PSA(个别+)。术后诊断:⑴ 戈谢氏病(I 型); ⑵ 脾脏肿大; ⑶ 脾功能亢进;⑷ 癫痫。
患者术后一般情况可,生命体征平稳,白细胞、红细胞及血小板均上升,特别是血小板明显增高,术后3 d血常规:白细胞11.60×109/L,红细胞4.32×1012/L,血红蛋白99 g/L,血小板162×109/L;术后1 周血常规:白细胞10.56×109/L,红细胞5.48×1012/L,血红蛋白102 g/L,血小板:62×109/L。由于经济原因,患者拒绝检测β- 葡萄糖脑苷脂酶水平、基因突变位点及使用酶替代疗法,患者于术后第5 天出院。术后 1 个月患者返院复查:血常规:白细胞8.67×109/L,红细胞4.67×1012/L,血红蛋白122 g/L,血小板362×109/L。在随后的3 个月的电话随访中,患者自述可以完成一些较轻的家务活,当地医院的血常规检查提示血小板轻度增高。
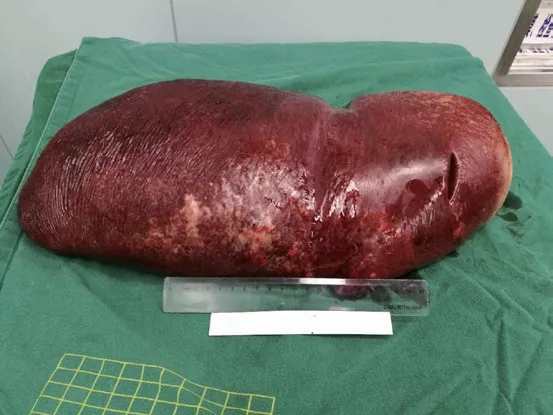
图1 完整切除的脾脏
讨论戈谢氏病(Gaucher's disease,GD)又称葡萄糖脑苷脂沉积病,是溶酶体贮积病(lysosomal storage disease,LSD)中最常见的一种,为常染色体隐性遗传病。神经类脂是神经鞘膜和细胞膜的重要组成部分。神经氨基醇与脂酸结合成神经酰胺,后者与葡萄糖分子结合成为葡萄糖脑苷脂,存在于集体的各组织中。GD 的病因主要是由于葡萄糖脑苷脂酶(GBA)编码的结构基因突变,导致编码的β- 葡萄糖脑苷脂酶缺乏,使得葡萄糖脑苷脂在肝、脾、骨骼、骨髓、肺、脑组织、淋巴结等器官的单核- 巨噬细胞中蓄积,形成典型的戈谢氏细胞[1-2]。该病在世界各地均有发病,不同群体中发病率有很大差别,以北欧犹太人的发病率较高,约1/450,而在犹太人群以外发病率约为1:10 万[3]。

图2 术后病理 A:HE 染色(×200);B:CD68(×200) ;C:PAS(×400)
GD 的临床表现包括贫血、血小板减少、肝脾肿大、骨骼疾病、神经病变等[4-6]。根据神经系统症状,临床上GD 通常分为三种表型。I 型(慢性非神经型)是迄今为止最常见的亚型并且不具有任何神经系统表现,任何年龄均可起病,主要表现为肝脾肿大、贫血、血小板较少,晚期可出现四肢骨痛,甚至出现病理性骨折,小儿患者其发育常受影响。此型GD 进展缓慢,脾切除后可长期存活,预后最好。II 型(急性神经型)是疾病的急性形式,患儿自出生即可有肝大、脾大,神经病变突出,病程短暂,多早期死亡,通常为2 岁,预后差。 III 型(亚急性神经型)的特征是神经系统症状较晚发作,常于2 岁至青少年期发病,临床病程较长,早期表现与I 型相似,逐渐出现神经系统受累表现,患者常有惊厥、共济失调、肌张力增高等表现,伴发育迟缓、智力落后。此型GD 患者多由于后期神经症状较重,死于并发症,近年来由于酶替代治疗的应用,预后有了较大的改观[6-8]。
GD 的诊断通常可以根据肝脾肿大或有中枢神经系统症状、骨髓涂片、酶学检查、基因诊断及病理活检进行,但最准确的是基于β- 葡萄糖脑苷脂酶的检测及DNA 检测[9]。 ⑴ 酶学检查:测定外周血白细胞中β- 葡萄糖脑苷脂酶活性。β- 葡萄糖脑苷脂酶低于15% 是诊断性的。酶学检查敏感、特异,而且侵袭性小,可以在产前进行[10]。⑵ 基因诊断:目前已鉴定出200 多个不同的突变型GBA 等位基因,其中最常见的是N370S、L444P、84GG 和IVS2,约占致病等位基因的90%。突变分析对疾病进展有一定的预测价值,如N370S 突变纯合子的患者,预后较好,一般无神经系统症状。但基因型与临床表型之间没有确定的联系[10-11]。 ⑶ 骨髓涂片:在片尾、边缘处可找到戈谢细胞,这种细胞体积大、直径约20~80 μm,有丰富胞浆,内充满交织成网状或洋葱皮样条纹结构,有1 个或数个偏心核;糖原染色阳性[10]。 ⑷ 其他:血常规可提示三系减少,B 超检查可提示肝脾肿大,X 线检查可提示广泛性骨质疏松等[12]。
GD 需与尼曼- 皮克病、GM1 神经节苷脂贮积症、GM1 神经节苷脂贮积症等病相鉴别,可通过骨髓涂片查看有无戈谢细胞相鉴别。对于GD 与具有戈谢细胞的疾病鉴别,如慢性粒细胞白血病、重型珠蛋白生成障碍性贫血及慢行淋巴细胞白血病,可通过患者的症状、β- 葡萄糖脑苷脂酶的测定相鉴别。
GD 虽无特效疗法,但近20年来GD 的治疗取得了突破性进展。尤其是酶替代治疗(enzyme replacement therapy,ERT),它被认为是第一线疗法,ERT 已经被证明可以改善血液学参数(血红蛋白浓度和血小板计数)、器官肿大和骨骼受累[13-14]。但肺部及骨骼的一些病变是不可逆转的,如骨坏死、骨纤维化和溶解性病变,所以早期诊断、早期治疗显得尤为重要。对于ERT 不适合的轻、中度GD1 患者,考虑用葡萄糖苷酰鞘氨醇的结构类似物进行底物抑制剂治 疗(Substrate reduction therapy,SRT)作为二线治疗选择[15-16]。由于ERT 花费较高,虽为一线用药,但很多患者仍无力支付,只能对症治疗,包括营养支持、输全血或红细胞、止痛、脾切除、免疫支持治疗等。此外药理性分子伴侣治疗、基因治疗剂细胞治疗也有了新进展,但仍在探索阶段[2]。随着脾脏逐渐增大,严重影响生活,患者常伴有脾功能亢进,切脾可缓解脾亢症状、预防脾破裂、改善出血。本例患者从发现脾大至今足足13年,此次决定来院就诊行手术治疗是由于严重影响生活,在经济欠发达的地区,很多患者常常因为经济原因无法第一时间得到治疗,本例患者在5 余年前便已明确诊断,但由于酶替代治疗价格昂贵,患者未能得到更早治疗。目前术后患者一般情况良好,生活质量得到了提高,规律随诊中。
由于GD 以多器官受累为特征,因其缺乏特异性往往使GD 的诊断延迟,加之目前只有北京、上海、武汉及广州四个中心才能检测溶酶体贮积症的酶活性,使得许多远离测试中心的GD 患者直到出现典型症状、体征时才能得到确诊,这使得大多数GD患者错过了最佳的治疗时机。本例患者也是多年辗转全国多地求诊,花了近8年时间才明确诊断。
猜你喜欢
健康体检与管理(2022年2期)2022-04-15 01:33:37
中国宝玉石(2021年5期)2021-11-18 07:34:50
中老年保健(2021年8期)2021-08-24 06:22:38
人人健康(2017年19期)2017-10-20 14:38:31
临床医药文献杂志(电子版)(2017年11期)2017-05-17 04:48:47
当代医药论丛(2017年22期)2017-04-12 06:29:43
中国卫生标准管理(2015年15期)2016-01-15 02:58:43
中国卫生标准管理(2015年10期)2015-01-27 09:31:56
云南畜牧兽医(2014年2期)2014-02-28 21:25:16
河南医学研究(2014年4期)2014-02-27 14:52:23
