
Characteristics and advantages of adenoassociated virus vector-mediated gene therapy for neurodegenerative diseases
2019-03-15 05:50YuanQuYiLiuAhmedFayyazNoorJohnathanTranRuiLi
中国神经再生研究(英文版) 2019年6期
Yuan Qu, Yi Liu, Ahmed Fayyaz Noor, Johnathan Tran, Rui Li,
1 Department of Hand Surgery, the Second Hospital of Jilin University, Changchun, Jilin Province, China
2 Department of Orthopedics, Qilu Hospital of Shandong University, Jinan, Shandong Province, China
3 Department of Chemistry, University of Massachusetts Lowell, Lowell, MA, USA
4 Department of Premedical and Health Studies, Massachusetts College of Pharmacy and Health Sciences, Boston, MA, USA
Abstract Common neurodegenerative diseases of the central nervous system are characterized by progressive damage to the function of neurons, even leading to the permanent loss of function. Gene therapy via gene replacement or gene correction provides the potential for transformative therapies to delay or possibly stop further progression of the neurodegenerative disease in affected patients. Adeno-associated virus has been the vector of choice in recent clinical trials of therapies for neurodegenerative diseases due to its safety and efficiency in mediating gene transfer to the central nervous system. This review aims to discuss and summarize the progress and clinical applications of adeno-associated virus in neurodegenerative disease in central nervous system. Results from some clinical trials and successful cases of central neurodegenerative diseases deserve further study and exploration.
Key Words: nerve regeneration; central nervous system; gene therapy; neurodegenerative disease; viral vector;adeno-associated virus; Alzheimer's disease; Parkinson's disease; Huntington's disease; amyotrophic lateral sclerosis;spinal muscular atrophy; neural regeneration
Introduction
The central nervous system is a complex and delicate system where many of the pervasive disease processes arise. These diseases include a broad range of pathological states and can affect global or local metabolism and function (Simonato et al., 2013). Neurodegenerative diseases, which commonly occur in the central nervous system, are defined by progressive nervous system dysfunction associated with atrophy of the nervous structures. The altered maintenance of proteostasis is considered as a general feature among neurodegenerative diseases (Gerakis and Hetz, 2018). Prototypical neurodegenerative diseases in the central nervous system include Parkinson's disease (PD), Alzheimer's disease (AD), amyotrophic lateral sclerosis and Huntington's disease (Lin and Beal, 2006). In general, the most available therapeutic approaches have not been proven as an effective treatment for neurodegenerative diseases in the central nervous system.This is due to the lack of viable long-term delivery of therapeutic drugs to the central nervous system. Furthermore,the blood-brain barrier restricts the access of many systemic treatments and those that do pass often have effects on non-target areas compromising the widespread delivery of therapeutic drugs to the central nervous system (Lykken et al., 2018). Thus, there is a necessity for more effective strategies and therapies to be explored.
Currently, gene therapy has been developed as a novel strategy for treating both genetic inherited and acquired neurodegenerative diseases in the central nervous system. It can deliver a transgene to supply gene products that will permanently restore the missing functions or introduce a therapeutic gene to cells of the target tissue (Piguet et al., 2017). Both viral and non-viral vectors are commonly used vehicles for gene therapy (Bangde et al., 2017). In comparison with viral vectors, the instability and rapid clearance of non-viral vectors still need further resolution (Yin et al., 2014).
The viral vector-mediated gene therapy, which includes the vectors of lentiviruses, adenovirus, herpes simplex virus, vaccinia virus and adeno-associated virus (AAV), can deliver therapeutic genes directly to the central nervous system (Kotterman et al., 2015). However, the efficiency and safety of gene therapy are very dependent on the vectors.Many studies and developments have shifted toward the advancement of innovative viral vectors that could combine low genotoxicity and immunogenicity delivery with greater efficiency. Among these vectors, adeno-associated virus was regarded as a promising technological advance (Murlidharan et al., 2014).
The structure of AAV involves one single-stranded DNA with an inverted terminal repeat as the viral genome and one protein capsid (Cassinotti et al., 1988; Lentz et al., 2012;Pillay et al., 2016). Generally, it can package DNA to a size limit of less than 4.7 kb (Colella et al., 2018). AAV vectors have the ability to transduce distinctive cells or tissues by packaging non-genomic DNA that is not related to any central nervous system or neurodegenerative disease (Agbandje-McKenna and Kleinschmidt, 2011). AAV also combines low immunogenicity and less pathogenicity with long-lasting transgene expression in clinical applications. Some serotypes of AAV can cross the blood-brain barrier with ease.With an AAV2 capsid, the vector has been shown to target cerebral vascular endothelial cells specifically (Chen et al.,2009; Gray et al., 2010; McCown, 2011). After systemic ad-ministration with the AAV9 serotype, high expression was observed in the great mass of cerebral regions involving the substantia nigra par reticulata, hippocampus, cerebellum,motor cortex and cervical spinal cord (McLean et al., 2014).
The distinct structures of AAV-mediated gene therapy have allowed novel treatments of neurodegenerative diseases in the central nervous system, particularly with recombinant AAV. To analyze the application and related function of AAV, this review investigates the past, present and future statuses of AAV for the treatment of neurodegenerative diseases in the central nervous system. We performed literature searches by using PubMed, Scopus, Embase and Web of Science databases with no language restrictions. The time period ranged from July 1, 2005 to May 31, 2018. Search terms include “central nervous system” or “gene therapy” or“neurodegenerative disease” or “viral vector” or “adeno-associated virus” or “Alzheimer's disease” or “Parkinson's disease” or “Huntington's disease” or “amyotrophic lateral sclerosis” or “spinal muscular atrophy” or “Canavan disease”or “Metachromatic leukodystrophy”. Eligibility criteria were as follows: All articles and reviews regarding AAV-mediated gene therapy for neurodegenerative diseases or any specific neurodegenerative disease of the central nervous system were included. In addition, we searched the reference lists of the retrieved papers for relevant articles. The full texts of the relevant articles were read to extract information on the topic of interest (summarized in Table 1). The procedures used generalized AAV-mediated gene therapy for the different neurodegenerative diseases of the central nervous system as follows.
Alzheimer's Disease
AD is one of the age-related neurodegenerative disorders and is accompanied by the progressive loss of cognitive function and impaired learning ability. It is associated with progressive deficiency in synapses and neurons of the cerebral cortex (Fol et al., 2016; Javidnia et al., 2017). The causes for most AD are still unknown, and several competing hypotheses have been proposed to elucidate the pathogenesis of the disease. In recent years, AAV has been used to test different hypotheses with the therapy having the advantages of long-term genome expression and fewer immune responses.The accumulated evidence has indicated that neuroinflammation is one of the main reasons for AD (Heppner et al., 2015; Onyango, 2018; Wang et al., 2018a). It was reported that interleukin-2, which is considered to act as an in flammation controller, could be packaged in an AAV vector and applied to alleviate AD in mice model (Saadoun et al., 2011). AAV-interleukin-2 was able to remodel the hippocampus by rescuing spine density and improving synaptic plasticity that led to progressive recovery of memory ability with only rare side effects (Alves et al., 2017). In the neuroin flammatory process, microglia, which highly express the pro-in flammatory molecule glia maturation factor, have been proven to play another important role (Cornejo and von Bernhardi, 2016; Crotti and Ransohoff, 2016; Colonna and Butovsky, 2017). It was found that glia maturation factor was expressed in various regions of AD brains. However,reduction of glia maturation factor could protect neuronal degeneration in PD mouse models. Raikwar et al. (2018)applied AAV-mediated gene therapy to downregulate glia maturation factor gene expression in the reactive microglial cell line. The progression of neuroin flammation was inhibited after genome editing, indicating that the downregulation of AAV-mediated glia maturation factor might be a potential target for AD treatment (Raikwar et al., 2018).
In the amyloid hypothesis of AD, the overexpression or accumulation of amyloids induces in flammation, leading to neuronal death (Hardy and Selkoe, 2002; Tong et al., 2018;Zhang et al., 2018). Therefore, the amyloid precursor protein whose proteolysis generates amyloid beta is considered as a potential target for AD treatment. Kiyota et al. (2015)found that CD74 could bind to amyloid precursor protein and inhibit amyloid beta processing. Subsequently, they showed that AAV-mediated CD74 expression decreased the production of amyloid beta and improved the brain function in mice (Kiyota et al., 2015). Some therapeutic enzymes delivered by AAV, such as asparagine endopeptidase and neprilysin 2, also achieved positive effects in an AD animal model. These enzymes could rescue the loss of synaptic functions and delay cognitive deficits by degrading amyloid beta (Sasmita, 2018). These results have inspired a new approach to treating AD.
The degeneration of the nucleus basalis of Meynert, which can cause cortical cholinergic deficits, is considered another feasible pathogenesis in AD. To delay or even stop degeneration of the nucleus basalis of Meynert in patients, a Phase II clinical trial (Identifier: NCT00876863) that delivered nerve growth factor intracerebrally by AAV2 was performed.The rationale for this trial was based on preclinical animal research that showed nerve growth factor deprivation led to the degeneration of nucleus basalis of Meynert cells.Conversely, exposure to nerve growth factor rescued or increased cholinergic function in the deprived nucleus basalis of Meynert neurons (Rafii et al., 2014; Hunsberger et al.,2016; Cummings et al., 2017). However, in the clinical trial,no benefit of AAV2-nerve growth factor group was observed compared with sham surgery at 24 months post procedure.Due to this unsatisfactory result, more accurate and specific gene targeting is needed (Ra fii et al., 2018).
For late-onset AD, apolipoprotein E (APOE) alleles have an effect on AD (Yi et al., 2014). One of APOE alleles,APOE4, which leads to the massive accumulation of amyloid beta in AD brain, can accelerate the likelihood of AD occurrence and reduce the age of onset. However, APOE2 serves a protective role in AD. In a mouse model, it was reported that AAV-mediated APOE2 delivery could alleviate symptoms of AD (Zhao et al., 2016). In addition, a Phase I clinical trial(Identifier: NCT03634007), which uses AAVrh.10 as the viral vector to deliver APOE2 to AD patients, is in process in 2018(Table 1).
Taken together, AAV-mediated gene therapy has been applied in various pathogenesis of AD. Even though the Phase II clinical trial (AAV2-nerve growth factor) did not achieve the expected goal, AAV vectors can still provide a safe means to deliver therapy for AD (Figure 1).
Parkinson's Disease
PD is another age and neuroin flammation related neurodegenerative disease caused by the progressive loss of dopaminergic neurons in the substantia nigra (Shi and Chen., 2017;Martinez and Peplow, 2018). Traditional anti-Parkinson agents, such as levodopa and dopamine agonists, are widely used in clinical treatment. However, as the disease progresses, these drugs become less effective and often lead to additional side effects. Lately, AAV-mediated gene therapy has been extensively explored for treating PD. It aims to rescue damaged neurons by delivering neurotrophic factors or rebuild functions by delivering key enzymes involved in dopamine synthesis and metabolism (O'Connor and Boulis,2015). There have been some interesting reports on clinical trials of gene therapy application in PD since 2013 (Table 1).
Neurturin is a member of the glial cell line-derived neurotrophic factor family of ligands. Some animal experiments showed that neurturin was able to rescue dopaminergic neurons and improve motor function (Gash et al., 1996;Emborg et al., 2009; Herzog et al., 2013). After preliminary clinical tests, a clinical trial was conducted using AAV2-mediated neurturin (Cere-120) gene delivery (Identifier: NCT00985517). In this Phase II trial, 51 advanced PD patients who received AAV2-neurturin-treated or sham-operated agent were randomly divided into two groups.The agent was administered directly into the putamen and substantia nigra. However, 15 months post-administration,clinical outcomes were evaluated and showed that there was no improvement in AAV2-NRTN-treated patients. This result might be caused by the downregulation of neurturin and glial cell line-derived neurotrophic factor receptors in advanced PD patients (Warren Olanow et al., 2015).
Aromatic L-amino acid decarboxylase (AADC), the inhibitor of premature conversion of levodopa to dopamine,is another treatment for PD. One Phase I clinical trial(Identifier: NCT00229736) using AAV2-AADC to delay the decarboxylation of levodopa in nigrostriatal dopamine neurons indicated that AAV2-mediated AADC gene therapy was safe. Although AADC expression was stable over the four years in patients, the unified PD rating scale was improved only in the first 12 months (Christine et al., 2009;Mittermeyer et al., 2012). Recently, a magnetic resonance imaging-guided delivery system with convection enhanced delivery used in a non-human primate experiment showed a better effect of the treatment (Zeiss et al., 2017). Based on this, the intraputaminal infusion of AAV2-AADC by stereotaxic surgery was used in another Phase II clinical trial(Identifier: NCT02418598). This clinical trial is now recruiting, and the results should be interesting.
Glutamic acid decarboxylase (GAD) is considered another target in PD treatment. It is the rate-limiting enzyme for regulating gamma-aminobutyric acid production. Recently, a controlled, blinded Phase II trial that delivered AAV2-GAD by directly injecting to the subthalamic nucleus showed promising efficacy (Identifier: NCT00643890). AAV2-GAD gene therapy improved unified the PD rating scale motor score at six months. However, the improvement of patients in the treatment group did not exceed the existing treating methods, so the study was terminated (LeWitt et al., 2011).
Despite some unsatisfactory clinical trial results, the research and exploration of PD treatment has not stopped.Oh et al. (2015) demonstrated that combined transcription factors, Nurr1 and Foxa2, delivered by an AAV vector could protect midbrain dopamine neurons and improve motor behaviors in mice. Another repulsive guidance molecule a, which regulates axon guidance and neuronal apoptosis,could also be a target. Repulsive guidance molecule a is upregulated markedly in the substantia nigra of PD patients.To verify the effect of repulsive guidance molecule a on dopaminergic neurons, repulsive guidance molecule a was delivered to the midbrain of mice using an AAV vector. It was reported that repulsive guidance molecule a in fluenced the PD process when analyzed at molecular, anatomical and behavioral levels (Oh et al., 2015; Korecka et al., 2017).
Huntington's Disease
Huntington's disease is an inherited gene disorder, which leads to progressive degeneration/death of striatal neurons (Ramaswamy and Kordower, 2012; Boussicault et al.,2016). Researchers are dedicating time and effort to clarify the pathogenesis of Huntington's disease. Korean scientists showed that an AAV vector, serotype DJ, could generate a novel juvenile-mouse model of Huntington's disease (Jang et al., 2018). After treatment with AAV-DJ, the behaviors of mice in the model replicated the dysfunction of motor neurons and neurodegeneration observed in Huntington's disease. The changes in animal behavior were accompanied by extensive neural cell apoptosis, the upregulation of inflammatory cytokines and activation of glia. This model illustrated that the use of AAV vector could help understand the mechanisms in the Huntington's disease striatum and explore therapeutic strategies for Huntington's disease (Jang et al., 2018).
One AAV vector-mediated gene therapy in a Huntington's disease animal model showed robust therapeutic effects(Dufour et al., 2014; Vagner et al., 2016). It is reported that Huntington's disease transgenic sheep, with human mutant HTT, were conducted for Huntington's disease study. When AAV9-mediated miRNA, which targeted exon 48, was delivered to Huntington's disease sheep striatum, human mutant(m) HTT mRNA and 50—80% proteins encoded by mutant Huntington gene were eliminated six months post injection.Moreover, the Iba1-positive microglia could be detected at the same level as the control group (P fister et al., 2018). In another experiment, a SIRT3 gene delivered by AAV vector protected Huntington's disease mice from neurodegeneration by blocking mitochondrial oxidative stress and maintaining neuronal bioenergy (Cheng et al., 2016). In addition,AAV5 noticeably reduced the mutant Huntington mRNA and protein when delivering the Huntington-lowering gene in minipigs. These results illustrate the safety and effective-ness of AAV vectors in treating Huntington's disease. Figure 1 summarizes research to date on the brain areas associated with each degenerative disease and the AAV vectors used to apply therapies. The laboratory and preliminary clinical research on Huntington's disease has reached the stage where treatment could start to be applied clinically (Evers et al.,2018).
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis, also known as Lou Gehrig's disease, is a neuromuscular disease. Amyotrophic lateral sclerosis is characterized by degeneration of the motor cortex, brainstem, and spinal cord, along with atrophy in motor neurons and muscles. The incidence of amyotrophic lateral sclerosis is only 4.3 per 100,000 in the United States(P fister et al., 2018). However, amyotrophic lateral sclerosis is a disease which leads to paralysis and premature death(Bertram and Tanzi, 2005; Yamashita et al., 2013). Recently,AAV9-mediated human insulin-like growth factor 1 was applied to the amyotrophic lateral sclerosis mouse model. This research showed that AAV9-mediated insulin-like growth factor 1 therapy decreased the amount of apoptotic motor neurons and prolonged the lifespan of amyotrophic lateral sclerosis mice. The pathway has been associated with the blocking of phosphorylated p38 mitogen-activated protein kinase and c-Jun-N terminal kinase in the spinal cord (Wang et al., 2018b).
The suppression of toxic mutant superoxide dismutase 1 (SOD1) has been considered one of the most promising treatments for amyotrophic lateral sclerosis for the last few decades. Biferi et al. (2017) reported that AAV10, packaged in a modified U7 small nuclear RNA, reduced the mutant SOD1 level through regulating exon skipping of the mutant SOD1 pre-mRNA in an amyotrophic lateral sclerosis mouse model. The treatment prevented the loss of body weight, relieved neuromuscular function damage and led to an increase in survival of SOD1-G93A mice. Li et al. (2017)pointed out that the injection speed of AAV10 affected the transduction of the spinal cord and dorsal root ganglia. If the injection speed was decreased, the transduction efficiency was increased (Biferi et al., 2017; Li et al., 2017). These encouraging results of the application of AAV in amyotrophiclateral sclerosis should inspire further research in patients(Figure 1).

Table 1 AAV-mediated gene therapy in clinical trials for neurodegenerative diseases in the central nervous system
Spinal Muscular Atrophy
Spinal muscular atrophy is an inherited neuromuscular disorder caused by motor neuron degeneration in the anterior horns of the spinal cord and brain stem (Parente and Corti,2018). Spinal muscular atrophy leads to progressive weakness and atrophy of muscle. Respiratory muscle failure is usually the fatal factor for spinal muscular atrophy patients.Spinal muscular atrophy arises from the loss of survival motor neuron 1 (SMN1) gene, which encodes the essential protein, SMN protein, for alpha motor neurons (Kariyawasam et al., 2018). In spinal muscular atrophy patients the SMN1 gene is replaced by a mutated identical gene, SMN2, which encodes an unstable and partially functional protein due to the splicing of exon 7 (Scoto et al., 2018). The clinical severity of the disease is associated with the onset age and the number of SMN2 copies. Based on this, spinal muscular atrophy is classified as four phenotypes (Lefebvre et al., 1997;Parsons et al., 1998; Messina, 2018; Tizzano and Zafeiriou,2018). Since the pathological mechanism of spinal muscular atrophy has been explained, many emerging strategies focus on the regulation of the deficiency gene. Recently, the application of AAV9 to deliver a functional SMN1 gene has been considered as a promising candidate for treating spinal muscular atrophy. Firstly, the studies in mice have verified that AAV9-AVXS-101 achieves the expression of SMN1 gene in the spinal cord after an intravenous administration. Moreover, the pathological symptoms of the spinal muscular atrophy mice have also been corrected (Foust et al., 2010; Passini et al., 2010; Valori et al., 2010). After verification in the laboratory, some clinical trials have been initiated (Table 1).The first Phase I trial (Identifier: NCT02122952) that tested the safety and efficiency of intravenous delivery of AAV9-AVXS-101 to 15 spinal muscular atrophy type I infants has been completed. The outcomes showed that the 15 patients who received a single intravenous administration of AAV9-AVXS-101 were all alive (an age of at least 20 months)along with improved motor function and increased CHOP INTEND scores up to August 2017 (Mendell et al., 2017).These results encourage great confidence in curing spinal muscular atrophy. In the meantime, other AAV-mediated spinal muscular atrophy clinical trials are being initiated as part of a wide application of AAV gene therapy. The latest Phase III trial (Identifier: NCT03505099) characterized by recruiting patients with multiple phenotypes was just posted in 2018.
Canavan Disease
Canavan disease is a neurodegenerative disease defined by diffused spongiform white matter degeneration in the brain,dysmyelination and intramyelinic oedema along with consequent deterioration of psychomotor development (Matalon and Michals-Matalon, 1999; Kumar et al., 2006; Hoshino and Kubota, 2014). The pathology of Canavan disease is caused by inactive aspartoacylase, which leads to the toxic accumulation of N acetylaspartate. Currently, traditional treatments for Canavan disease include the application of lithium, glyceryl triacetate, triheptanoin and oral N acetylaspartate administration (Janson et al., 2005; Madhavarao et al., 2009; Arun et al., 2010; Baslow and Guilfoyle, 2013;Francis et al., 2014). However, it is evident that an efficient gene therapy for Canavan disease would be an ideal treatment if begun at an early age. A Phase I clinical trial which delivered AAV2-aspartoacylase intraparenchymally to Canavan disease patients has been completed (Table 1). A long-term follow up of gene delivery in 13 Canavan disease patients was published by Leone et al. (2012). Their results showed the treatment was well tolerated and no serious side effects occurred. The therapeutic benefits were decreased accumulation of brain N acetylaspartate; delayed progressive brain atrophy; and less frequent seizures. Neurological examination showed significant motor function improvements in the younger patients treated for Canavan disease,suggesting the probable advantage of early-onset therapeutic intervention (McPhee et al., 2006; Leone et al., 2012; Ahmed and Gao, 2013).
Metachromatic Leukodystrophy
Metachromatic leukodystrophy is an inherited lysosomal disorder caused by deficiency of the enzyme arylsulfatase A.This results in excess accumulation of sulfatides in Schwann cells, oligodendrocytes, and brain neurons (van Rappard et al., 2015). Currently, there are several promising treatments available for metachromatic leukodystrophy along with bone marrow transplants, enzyme-replacement therapy and AAV-mediated gene therapy directly to the central nervous system (Rosenberg et al., 2016). A Phase I/II clinical trial of intracerebrally delivering AAVRh10-arylsulfatase A to metachromatic leukodystrophy patients is in process (Table 1). This clinical trial follows previous successful AAV-mediated gene therapy experiments in mice (Piguet et al., 2012;Miyake et al., 2014). The safety and efficacy of AAVRh10-arylsulfatase A on early-onset metachromatic leukodystrophy are being assessed by clinical, neuropsychological, radiological, electrophysiological and biological parameters. It could become a promising method to treat metachromatic leukodystrophy patients.
Conclusion
In the past decades, AAV has been developed as a promising nonpathogenic vector of gene therapy to treat neurodegenerative diseases in the central nervous system. It can deliver the therapeutic gene to the central nervous system directly and provide long-term and functional correction of missing or mutated genes. The development of different serotypes and genetic targets for different cells could assist in the curing of neurodegenerative diseases stemming from various causes. A powerful example of AAV and its established achievements are illustrated in the successful curing of spinal muscular atrophy in young infants.
However, there have been some poor results in other trials using AAV. Some of the failures may be attributed to the lack of complete and thorough knowledge concerning the diseases rather than the errors arising from the use of AAV itself. The clinical application of AAV faces other challenges. The neutralizing antibodies against AAV are considered as a major obstacle. The pre-existing antibodies which can neutralize AAV have severely limited the broad use of AAV vectors in clinical gene therapy (Ortolano et al., 2012; Rapti et al., 2012). Another limitation for the application of AAV is target specificity. The receptors for different serotypes of AAV are still being explored. To date, AAVR and galactose could bind more specifically to AAV2 and AAV9 respectively, but these receptors are located in many organs in the human body (Madigan and Asokan, 2016; Pillay et al., 2016).This means that it is difficult to deliver AAV2 or AAV9 to unique cells or tissue after a systemic administration.
Other issues, such as optimal administration methods and manufacturing problems, need to be addressed. These efforts should accelerate and improve the clinical application of AAV vectors in each neurodegenerative disease of the central nervous system. AAV is a promising vector in the new application of gene therapy and is deserving of more opportunities for further research, exploration, and implementation.
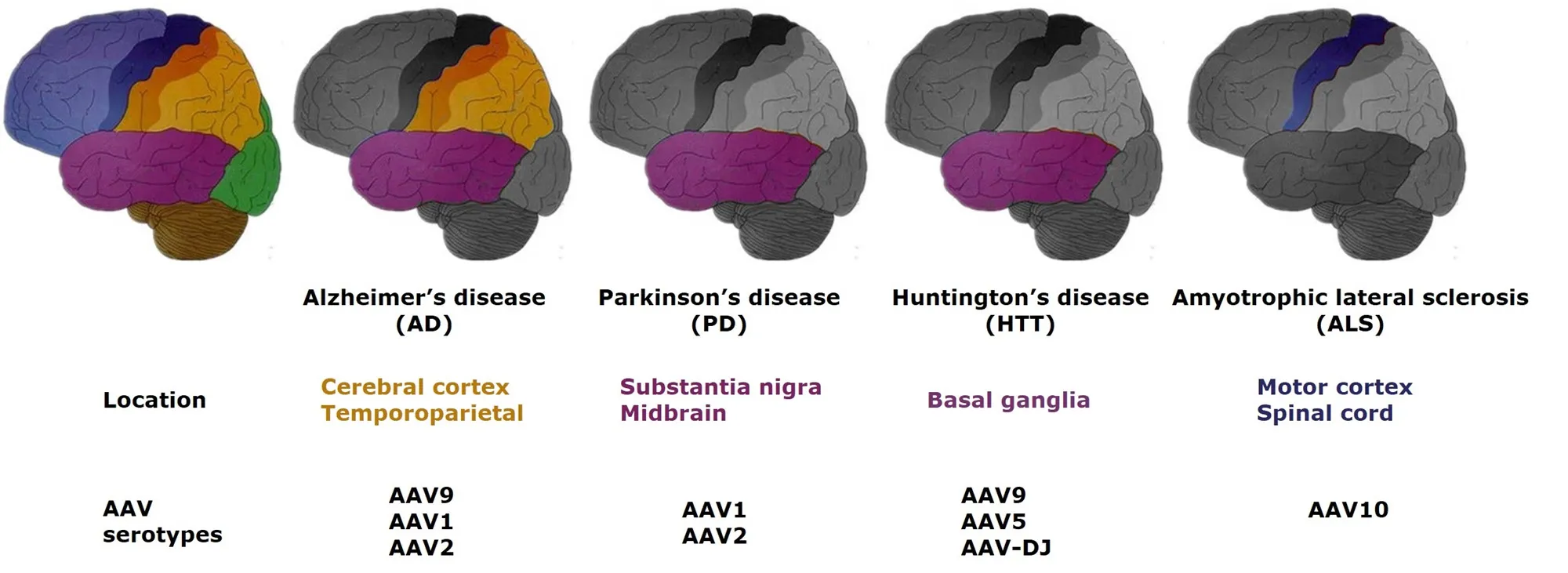
Figure 1 General locations of the main neurodegenerative diseases in the central nervous system and related AAV serotypes, which have been published.
Author contributions:Material collection and manuscript writing: YQ;review design, authorization and instruction: RL; writing procedures and literature search: YL and AFN; manuscript revision: JT. All authors approved the final version of the manuscript.
Conflicts of interest:The authors declare that they have no competing interest.
Financial support:None.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Panteleimon Giannakopoulos, University Hospitals of Geneva, Switzerland; Marvin Soriano-Ursua, Escuela Superrior de Medicina IPN, Mexico.
Additional file:Open peer review reports 1 and 2.
- 中国神经再生研究(英文版)的其它文章
- Muscle secretion of toxic factors,regulated by miR126-5p, facilitates motor neuron degeneration in amyotrophic lateral sclerosis
- Comparative study of microarray and experimental data on Schwann cells in peripheral nerve degeneration and regeneration: big data analysis
- Busting the myth: more good than harm in transgenic cells
- Gene expression changes in dorsal root ganglia following peripheral nerve injury: roles in in flammation, cell death and nociception
- Nicotinamide adenine dinucleotide phosphate oxidase activation and neuronal death after ischemic stroke
- Autophagy: novel insights into therapeutic target of electroacupuncture against cerebral ischemia/reperfusion injury