
Integrated network analysis of transcriptomic and protein-protein interaction data in taurine-treated hepatic stellate cells
2019-03-11 08:47XingQiuLiangJianLiangXiaoFangZhaoXinYuanWangXinDeng
Xing-Qiu Liang, Jian Liang, Xiao-Fang Zhao, Xin-Yuan Wang, Xin Deng
Abstract BACKGROUND Studies show that the antifibrotic mechanism of taurine may involve its inhibition of the activation and proliferation of hepatic stellate cells (HSCs). Since the molecular mechanism of taurine-mediated antifibrotic activity has not been fully unveiled and is little studied, it is imperative to use “omics” methods to systematically investigate the molecular mechanism by which taurine inhibits liver fibrosis.AIM To establish a network including transcriptomic and protein-protein interaction data to elucidate the molecular mechanism of taurine-induced HSC apoptosis.METHODS We used microarrays, bioinformatics, protein-protein interaction (PPI) network,and sub-modules to investigate taurine-induced changes in gene expression in human HSCs (LX-2). Subsequently, all of the differentially expressed genes(DEGs) were subjected to gene ontology function and Kyoto encyclopedia of genes and genomes pathway enrichment analysis. Furthermore, the interactions of DEGs were explored in a human PPI network, and sub-modules of the DEGs interaction network were analyzed using Cytoscape software.RESULTS A total of 635 DEGs were identified in taurine-treated HSCs when compared with the controls. Of these, 304 genes were statistically significantly up-regulated, and 331 down-regulated. Most of these DEGs were mainly located on the membrane and extracellular region, and are involved in the biological processes of signal transduction, cell proliferation, positive regulation of extracellular regulated protein kinases 1 (ERK1) and ERK2 cascade, extrinsic apoptotic signaling pathway and so on. Fifteen significantly enriched pathways with DEGs were identified, including mitogen-activated protein kinase (MAPK) signaling pathway, peroxisome proliferators-activated receptor signaling pathway,estrogen signaling pathway, Th1 and Th2 cell differentiation, cyclic adenosine monophosphate signaling pathway and so on. By integrating the transcriptomics and human PPI data, nine critical genes, including MMP2, MMP9, MMP21,TIMP3, KLF10, CX3CR1, TGFB1, VEGFB, and EGF, were identified in the PPI network analysis.CONCLUSION Taurine promotes the apoptosis of HSCs via up-regulating TGFB1 and then activating the p38 MAPK-JNK-Caspase9/8/3 pathway. These findings enhance the understanding of the molecular mechanism of taurine-induced HSC apoptosis and provide references for liver disorder therapy.
Key words: Taurine; Hepatic stellate cells; Differentially expressed genes; Liver fibrogenesis; Transcriptomic; Protein-protein interaction network
INTRODUCTION
Hepatic fibrosis is a pathological condition that may gradually develop into liver cirrhosis or even hepatocellular carcinoma (HCC) if left without effective treatment[1,2].Unfortunately, effective clinical therapies to halt disease progression are still lacking.In the development of liver fibrosis, the central event is that quiescent hepatic stellate cells (HSCs) become activated and then transdifferentiate into myofibroblast-like cells.The process of HSC activation may be the important target to halt the development of hepatic fibrosis, but it occurs quickly. Accordingly, researchers have focused on the regulatory pathways of HSC activation upon injurious stimuli by activating or repressing gene transcription, epigenetic regulation, and post-transcriptional control.To date, they have identified many target genes implicated in HSC activation,includingTGFB1, Foxf1, JunD, andC/EBP[3,4]. It is reported that deletion of oneFoxf1allele can slow down or inhibit HSC activation[5]. Similarly, knockout ofJunD, which encodes a functional component of the AP1 transcription factor complex[6], alleviated CCl4-induced hepatic fibrosisviathe reduction of activated HSCs and downregulation of hepatic TIMP-1 expression[7]. Furthermore, phosphorylation of the transcription factor C/EBP by RSK kinase promoted the survival of quiescent HSC[8].There could be no doubt that the regulatory pathways and key genes included in the process of HSC activation would be essential to develop therapeutic strategies against fibrogenic diseases.
Taurine, also known as 2-aminoethanesulfonic acid (C2H7NO3S), is a beta amino acid with a simple structure and mostly appears in the free state in organism. It plays a protective role in various cells and tissues[9]. It is reported that taurine can protect the liver against several forms of hepatic damage, including ischemia-reperfusion injury, hepatic carcinoma, and hepatic abnormality, which were demonstrated by animal experiments[10-15]. Furthermore, Miyazakiet al[13]investigated how taurine influences the hepatic fibrogenesis in rats or HSCs, and finally discovered that taurine could inhibit the proliferation of activated HSCs. In our previous studies, the methods of microculture tetrazolium and flow cytometry were performed to compare the apoptosis rate between taurine-treated and non-treated HSCs, and the results showed that taurine can inhibit cell proliferation and promote cell apoptosis significantly[16,17].Thus, supplementation with taurine should be considered a therapeutic approach to lessen the severity of liver injury and hepatic fibrosis. However, life is so complex that the therapeutic mechanism of any drug may involve a variety of genes and pathways in regulating biological networks. The molecular mechanism of taurine remains unclear, and therefore, it is difficult to use taurine for precision therapies in liver diseases. With the discovery and development of high-throughput research methods,the technology of microarray and bioinformatics give us an opportunity to analyze a number of genes related to complex refractory effects of traditional Chinese medicine.It is well known that the phenotype of a cell, ranging from the components to the functions, is ultimately up to its gene expression profiles. Analyzing the changes of gene expression profiles after treatment by medicine may help reveal their action mechanisms.
In the current study, we performed gene microarray and bioinformatics methods on taurine-treated human HSCs and control HSCs, which revealed differentially expressed genes (DEGs) between taurine-treated HSCs and control cells.Subsequently, the DEGs were subjected to the analysis of gene ontology (GO)function and Kyoto encyclopedia of genes and genomes (KEGG) pathway. And then,we further explored the interactions of DEGs in a human protein-protein interaction(PPI) network and sub-modulesviaCytoscape software. The overall goals were to provide therapeutic targets of taurine and to have an in-depth insight into the molecular mechanisms by which taurine protects the liver.
MATERIALS AND METHODS
Materials
Human HSCs (LX-2) were purchased from XiangYa Central Experiment Laboratory,Central South University, Changsha, Hunan Province, China. Taurine was provided by Yuanlong Pearl Co. Ltd., Beihai, Guangxi Zhuang Autonomous Region, China.Dulbecco’s minimum essential medium (DMEM) was obtained from Hyclone (Logan,UT, United States). Fetal bovine serum (FBS) was purchased from Biochrom AG(Berlin, Germany). Streptomycin sulfate and penicillin were supplied by North China Pharmaceutical, China. Distilled water was filtered through a Milli-Q system(Millipore, Bedford, MA, United States).
Culture and treatment of HSCs
HSCs were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin,and 100 U/mL streptomycin in an incubator at 37 °C and 5% CO2. The fresh culture medium was replaced every other day. When the cell density achieved approximately 80% confluence, cells were trypsinized and resuspended in DMEM at a density of 1 ×105/mL. For taurine-treated cells, the supernatant was discarded after centrifugation,and the cells were incubated for 48 h in DMEM containing 40 mmol/L taurine, while the control group was incubated in DMEM containing the same concentration of HSCs without taurine.
Illumina microarray chip analysis
Illumina Bead Chip (San Diego, CA, United S tates) was constructed as a gene expression microarray chip. Each slide (HumanHT-12_V4) contained 47323 DNA probes (887 controls, 46436 tests). Total RNA from HSCs was extracted with Trizol(Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. RNA samples, with an A260/A280 ratio ranging between 1.7 and 2.1, were subjected to denatured agarose gel electrophoresis for testing RNA integrity, followed by labeling through an Illumina®RNA Amplification kit (Ambion, Austin, TX, United States)according to the instructions of the manufacturers. RNA was converted to labeled cRNA and quantified as previously described[18].
Hybridization, washing, and detection were performed using the Illumina Gene Expression System Buffer Kit for HumanHT-12_V4 Bead-Chips (Illumina, San Diego,CA, United States) according to the manufacturer’s protocol. Briefly, an aliquot (11.3 μL) containing 850 ng cRNA was mixed with Hybridization Mix (22.7 μL), followed by incubating at 65 °C for 5 min. And then 34 μL reaction solution was transferred to Human HT-12_V4 Bead-Chip array, and incubated for 16 h at 55 °C with rotation.After hybridization, Bead-Chips were fully washed in supplied wash solutions 5-10 times on a shaker. For detection, Bead-Chips were transferred to a fresh wash tray containing streptavidin-Cy3 (1 μg/mL; Amersham Biosciences, Piscataway, NJ,United States) and rocked for 10 min, and then dried by centrifugation at 275gfor 4 min at 20 °C after washing. Arrays were scanned using Illumina Bead Array Reader and Bead Scan software, and subsequently analyzed using the software of Illumina Bead Studio Application (San Diego, CA, United States).
Microarray data acquisition and preprocessing
Raw data was obtained as “.IDAT” and “.SDF” format using Genome Studio software(Illumina, San Diego, CA, United States), and then imported into the R environment for further processing. Subsequently, quantile normalization was carried out in R using the lumi package with the Bioconductor open source software(http://www.bioconductor.org/).
Microarray data quality control and analysis
Text or excel files for each RNA hybridization were created by the Illumina®GenomeStudio Gene Expression Module (Version 1.0), and then analyzed in R3.2.5(http://www.R-project.org/). The Limma package[19](http://www.ncbi.nlm.nih.gov/pubmed/16646809) was used to perform background adjustment, summarization, and quantile normalization. Normalization was made using the robust multichip average (RMA) pre-normalization algorithm[20]. Data quality assessment was accomplished by using various quality control measures.Specifically, box plots are utilized to compare probe intensity levels among the arrays of the dataset. The median lines were not significantly different from each other after normalization. For each replicate array, gene expression ratios were generated by comparing each probe-set signal value from taurine-treated samples to that from control samples. DEGs were then identified by the Limma package with multiple testing correction using the Benjamini-Hochberg false discovery rate. The statistically significant DEGs were calculated using volcano plot analysis with the absolute value of log2 fold change (FC) (|log2FC| ≥ 1.5) and aP-value < 0.05. Moreover, DEGs were visualized by hierarchical clustering analysis.
Gene ontology function and Kyoto encyclopedia of genes and genomes pathway enrichment analysis
In order to explore their function, the DEGs with known identities or homologous sequences and known functional definitions were categorized using the Database for Annotation, Visualization and Integrated Discovery (https://david-d.ncifcrf.gov, ver.6.8) that is applied based on three categories of GO, namely, biological process (BP),molecular function (MF), and cell component (CC). Genes were excluded if they were only with expressed sequence tag identification or genomic coordinates that could not be annotated. Pathway enrichment analysis was performedviathe KEGG database to identify functional categories of statistically significant genes, which were defined as pathways exhibiting significantP-values (P< 0.05) with at least three overlapping genes. The biological networks were generated by comparing the input list of DEGs to a reference list from human databases.
PPI network
Human PPI networks were downloaded from the Human Protein Reference Database[21]. The human PPI network was then used to construct an interaction network of DEGs, and Cytoscape (http://cytoscape.org/, ver. 3.4.0) was used to mine the function modules. The sub-module in the interaction network was further explored using the MCODE plug-in in Cytoscape (the cut-off was set at 3).
Statistical analysis
Microarray data processing and normalization were analyzed in R3.2.5(http://www.R-project.org/), applying an RMA algorithm. Significance was calculated using at-test without multiple testing correction, selecting all transcripts with a minimum change in expression level of 1.5-fold together with aP-value less than 0.05. PPI network was analyzed using CytoNCA, which is a plugin of Cytoscape for network evaluation and topological analysis, including degree centrality,betweenness centrality, and closeness centrality, for hub gene screening. Proteins were defined as nodes, and PPI associations were defined as edges in the PPI network.
RESULTS
Effects of taurine on gene expression in HSCs
The results were consistent with our previous findings which demonstrated that taurine could up- and down-regulate gene expression in HSCs[22]. In the present study,635 genes showed significantly differential expression in taurine-treated HSCs as compared with the control under the threshold of |log2FC| ≥ 1.5 andP< 0.05. Of these, 304 (47.87%) were found to be up-regulated, while 331 (52.13%) were downregulated.
GO analysis of differential genes in taurine-treated HSCs
GO enrichment analyses were conducted on 304 differentially up-regulated and 331 down-regulated transcribed genes, which were classified into three main GO categories, including BP, MF, and CC. The distribution of annotated genes across different GO categories is shown in Table 1. The results indicated that DEGs were mainly involved in the MF of identical protein, calmodulin, protease, hormone,peptidoglycan, beta-amyloid, and calcium ion binding activity; olfactory receptor,steroid hormone receptor, and transmembrane signaling receptor activity; protein kinase, metalloendopeptidase, protein tyrosine kinase, metallopeptidase, serine-type peptidase, and transferase activity. In the light of the BP analysis, DEGs were involved in the processes of signal transduction, positive regulation of transcription from RNA polymerase II promoter, proteolysis, positive regulation of transcription,DNA-templated, negative regulation of cell proliferation, positive regulation of ERK1 and ERK2 cascade, and regulation of apoptotic process. In addition, DEGs were mainly located in the plasma membrane, integral component of plasma membrane,external side of plasma membrane, basement membrane, apical plasma membrane,integral component of membrane, extracellular region, presynapse, cell surface, and proteinaceous extracellular matrix.
Pathway analysis of DEGs in taurine-treated HSCs
In order to further identify the specific biological pathwa ys of differential genes associated with taurine treatment, the KEGG database was applied. As a result, 30 significantly enriched pathways with DEGs were identified (P < 0.05) and are listed in the Figures 1 and 2, which included mitogen-activated protein kinase (MAPK)signaling pathway, peroxisome proliferators-activated receptor signaling pathway,estrogen signaling pathway, Th1 and Th2 cell differentiation, cyclic adenosine monophosphate signaling pathway and so on (P< 0.05).
Analysis of the PPI network of DEGs
The interaction between protein and protein plays an important role in various BP. In the PPI network, the protein and the interaction are defined as the node and the edge,respectively. The present PPI network is comprised of 371 nodes and 882 edges(Figure 3). In this network, the ellipse nodes have higher degrees, the second is the triangle nodes, and the diamond nodes is the least. The ellipse nodes include 64 genes,such asJAK2, PRKACB, MAPK14, CYP19A1, RAP1A, TGFB1, ESR1, NPS, NR3C1,DNAH8, PIKFYVE, EGF, EGFR, BDNF, MMP9, andSRC. Of these, 32 were upregulated, and 32 down-regulated. The number of edges of each node is quite large(for example, 45 inSRC, 41 inPRKACB,MAPK14, andEGF, 40 inEGFR, and 33 inPIKFYVE). Thus, it indicated that the genes with more nodes are more important to the protective mechanism of taurine. Subsequently, we analyzed the network with a plugin of Cytoscape named MCODE, and obtained 11 clusters, which are showed in Table 2 and Figure 4.
DISCUSSION
In the present study, we explored the mechanism of taurine on human HSCs in more details based on the gene microarray data, identified the DEGs in the taurine-treated samples by a SAM-based method, analyzed the GO functions and KEGG pathways of DEGs, and further investigated the DEGs by interaction network and sub-module analysis.
The DEGs identified included matrix metalloproteinases (MMP2, MMP9, andMMP21), and their specific tissue inhibitor (TIMP3). It is generally recognized that metalloproteinases enhance cancer cell growth while their specific tissue inhibitor suppress. A collection of studies has indicated thatTIMP3could regulate the bioavailability of cytokines and growth factors to control inflammation and modulate the survival of cells in the liver. Studies targeting the hepatic fate, including liver regeneration[23], systemic inflammation[24], Fas-dependent hepatotoxicity[25], and high- fat diet-induced nonalcoholic fatty liver disease[26]or steatohepatitis[24], have confirmedTIMP3as a key controller of inflammation, cell death, and cell survival in the liver,again suggesting it as a tumor suppressor gene. It is noteworthy thatTIMP3is also found silenced in human cancers. Meanwhile, convincing evidence showed thatTIMP3loss slowed the progression of hepatic tumorigenesis and the development of malignancy despite eliciting heightened inflammation in response to carcinogen.Notably, gene deletion ofTIMP3could be dispensable by the immediate engagement of several tumor suppressor pathways to protect from carcinogen-induced HCC.These pathways include tumor necrosis factor signaling[27]. It will be seen from these that changes of these genes may be directly related to the protective effect of taurine against liver disease.

Table 1 Gene ontology enrichment results of differentially expressed genes
Moreover, Kruppel-like factor 10 (KLF10) is found to be up-regulated by taurine with an FC value of 2.83. It is a tumor suppressor, directly inhibits the expression of collagen I andPDGFRβ, and promotes HSCs apoptosis. Evidence indicates that haploinsufficiency ofKLF10led to fibrosis worsening in rats[28]. The up-regulation of KLFs by statins promoted the expression of the vasoprotective geneviaa KLF2-nitric oxide-guanylate cyclase-mediated paracrine mechanism[29]. It is suggested that taurine-increased apoptosis of HSCs may be associated with the function that upregulatesKLF10and then directly represses collagen I andPDGFRβexpression.

Figure 1 Kyoto encyclopedia of genes and genomes pathway analysis of the differentially expressed genes. The color represents different pathways. The larger area represents more DEGs enriched in the pathway. DEGs: Differentially expressed genes; cMAP: Cyclic adenosine monophosphate; MAPK: Mitogenactivated protein kinase; PPAR: Peroxisome proliferator-activated receptor.
Immune regulation, one of the cellular events in injured/inflamed hepatic tissue microenvironment, is also well known to directly lead to the activation of HSCs in a etiology-specific or -independent manner[30]. Investigation into the divergent immunomodulatory activities of quiescent and activated HSCsin vitrohas revealed that activated HSCs induce T-cell hyporesponsiveness[31]. The identified DEGs also include pro-fibrogenic inflammatory chemokines and their receptors such as C-X3-C motif chemokine receptor 1, which could increase activated HSC apoptosis[32]. These processes are fundamental to promote HSC apoptosis and the action mechanism of taurine may be directly associated with these regulation processes.
Based on the BP analysis of GO function, it is demonstrated that the DEGs were mainly involved in negative regulation of the cell proliferation, epidermal growth factor receptor signaling pathway, and regulation of apoptotic process. Several important pathways have been identified, including the MAPK signaling pathway,calcium signaling pathway, GnRH signaling pathway, and inflammatory mediator regulation of TRP channels. The MAPK signaling pathway is an intracellular signaling transduction pathway, including extracellular signal regulated kinase (ERK), Jun N-terminal kinase (JNK), p38, and ERK-5, and its major function is to control gene expression in response to cell proliferation, differentiation, and migration. In this intracellular signaling transduction pathway, JNK and p38 MAPK family members take effects in a cell context- and cell type-specific manner to integrate signals that affect proliferation, differentiation, survival, and migration[33]. Indeed, previous studies showed that ERK, JNK, and p38 MAPK regulate the activation of HSCs during the development process of liver fibrosis[30]. Moreover, it is reported that JNK may be involved in regulating activated HSC apoptosis by death receptors DR4/DR5 and pro-apoptosis factor CHOP[34]. JNK activity is also affected by inhibition of NF-κB,which may result in an increase of cell death and cytokine-driven compensatory proliferation[35]. Furthermore, Al-Azayzihet al[36]studied howTGFB1affected the cancer cell survival and proliferation, and found thatTGFB1decreased cell viability and induced cancer cell apoptosisviaactivation of the p38 MAPK-JNK-Caspase9/8/3 pathway. It is well known thatTGFβis an inhibitor of epithelial and lymphoid cell proliferation, and therefore considered to have an inhibitory effect on tumor. It is also included in the identified DEGs and was up-regulated by taurine with an FC value of 2.33 (P< 0.01). The conclusion was that taurine promotes the apoptosis of HSCsviaup-regulating TGFB1 and then activating the p38 MAPK-JNK-Caspase9/8/3 pathway.

Figure 2 Heatmap of enriched terms across the differentially expressed genes, colored by P-values. MAPK: Mitogen-activated protein kinase.
The PPI sub-network consisted ofVEGFB, EGF, BDNF, TGFB1, FIGF, TIMP3, FGB,EGFR, F7, MMP9, MMRN1, andTCOF1. The GO function analysis demonstrated that these DEGs are mainly involved in positive regulation of protein kinase B signaling,ERK1 and ERK2 cascade, MAP kinase activity, MAPK cascade, cell migration, and cell proliferation based on analysis of the BP. Meanwhile, it was also shown that these genes are mainly located on the plasma membrane. Furthermore, the KEGG pathway analysis indicated that these genes are mainly included in the MAPK signaling pathway, FoxO signaling pathway, Focal adhesion, Rap1 signaling pathway, Ras signaling pathway, and PI3K-Akt signaling pathway. VEGF is a member of PDGF/VEGF family that is related to the physiology of endothelial cells and responsible for blood vessel formation.VEGFis differentially spliced to form two isoforms:VEGFandVEGFb. WhereasVEGFstimulates angiogenesis,VEGFbis antiangiogenic[37]. Multiple factors can lead to up-regulation ofVEGFexpression,including hypoxia, oncogenic stimuli, and growth factors. The increasedVEGFexpression induces PI3K- and MAPK-dependent stimulation ofHIF-1α. In addition to provoking autophagic-mediated HSC activation,HIF-1αfeeds back to further stimulateVEGFproduction and continue the cycle[38]. EGF receptor is different between activated HSCs and quiescent HSCs. In the process that quiescent HSCs transdifferentiate into activated HSCs, ECM molecules would be secreted to accumulate and form scar tissue in the space of Disse, therefore EGF receptor is overexpressed and phosphorylated in activated HSCs. Pharmacological inhibition of EGF receptor could suppress hepatic fibrosis and HCC nodules[39]. In the present study, EGF is down-regulated. In general, the above findings suggested thatVEGFB,EGF, BDNF, TGFB1, FIGF, TIMP3, FGB, EGFR, F7, MMP9, MMRN1, andTCOF1play key roles in the action mechanism of taurine.
The analyses described here provide some basis for a better understanding of the molecular mechanism of taurine and for the development of treatment strategies for liver disorders. However, further experiments are required for validation. We would conduct the validation experiments in the following studies, including the differentially expressed genes and the p38 MAPK-JNK-Caspase9/8/3 pathway by using blockersviathe method of PCR and Western blot.
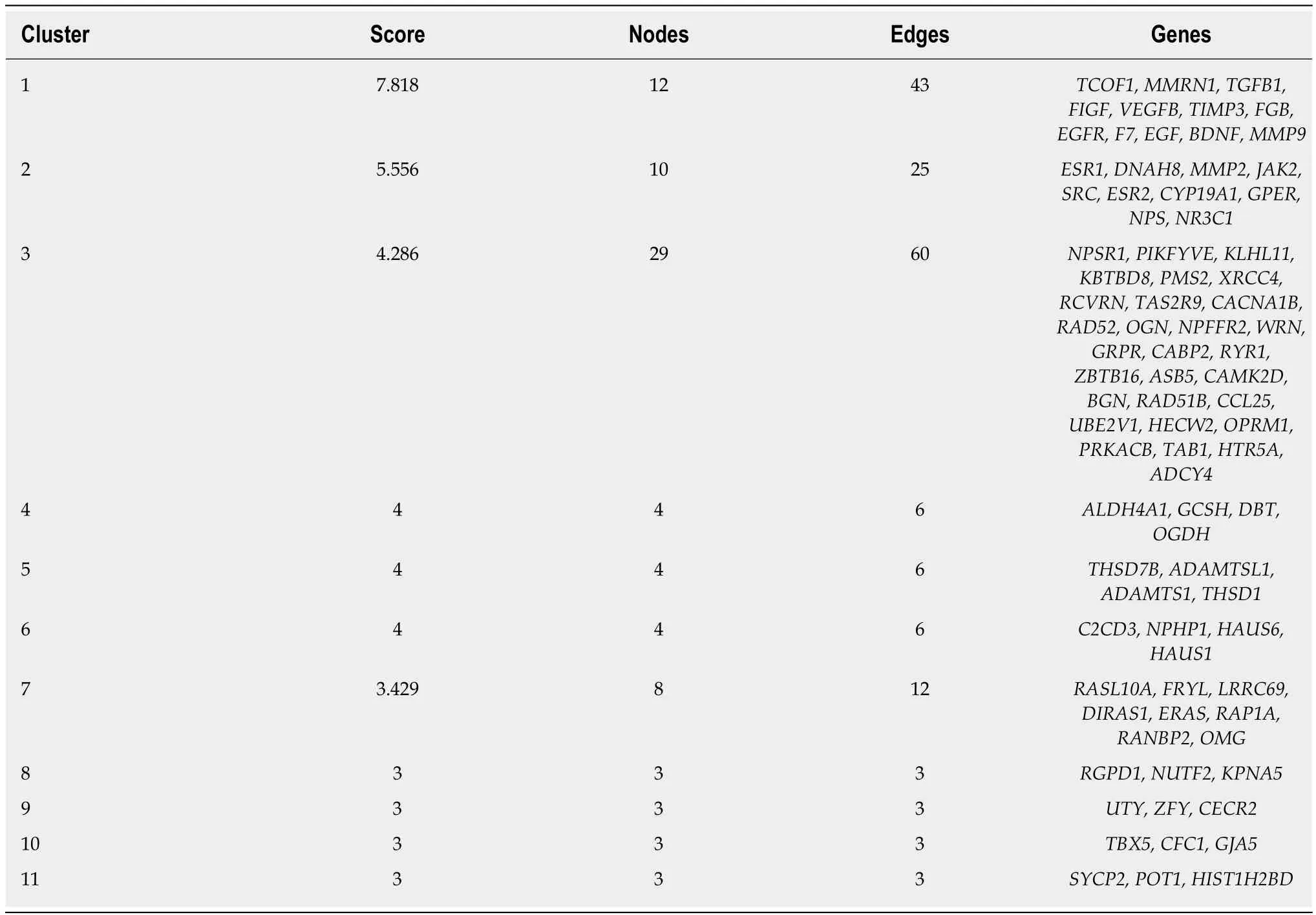
Table 2 Cluster of differentially expressed genes in the protein-protein interaction network
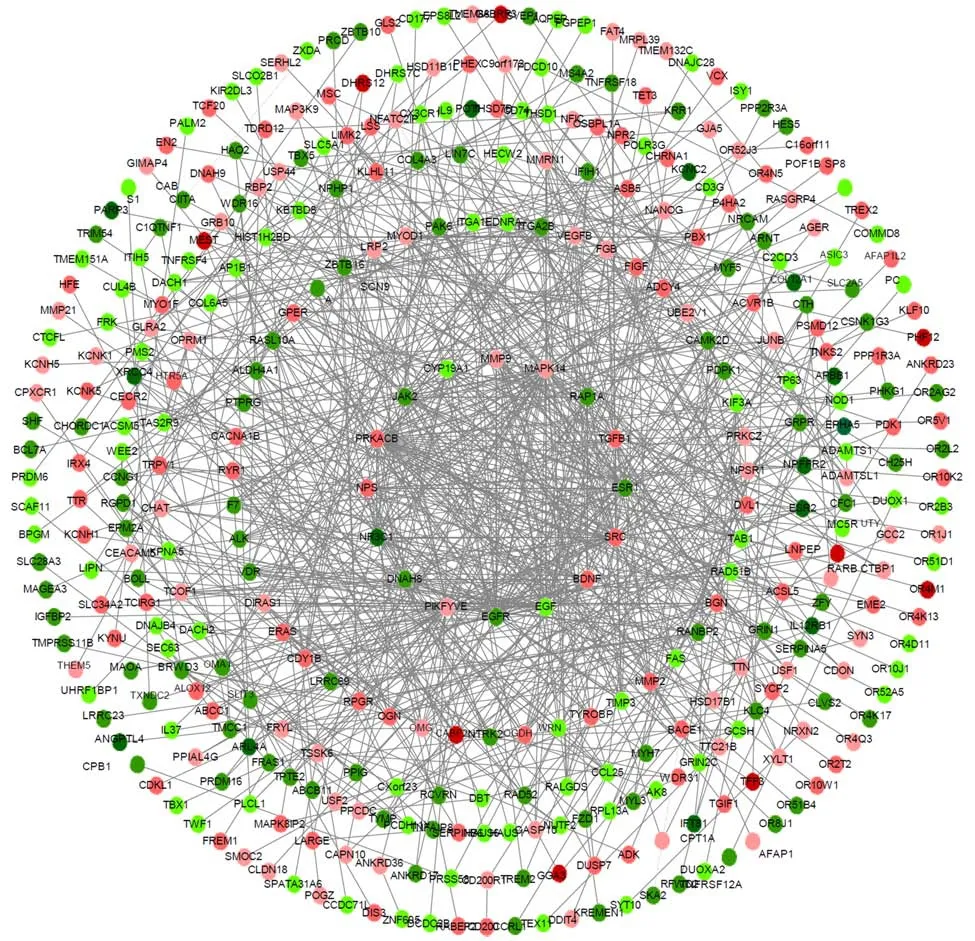
Figure 3 Protein-protein interaction network of differentially expressed genes in taurine-treated hepatic stellate cell samples. The nearer the center, the higher the degrees; red coloring represents up-regulated genes; green coloring represents down-regulated genes; purple coloring represents genes from database;the shade is related to the change fold (darker colors indicate more apparent differences).

Figure 4 Functional sub-modules in the protein-protein interaction network of differentially expressed genes in the taurine-treated hepatic stellate cells.Red coloring represents up-regulated genes; green coloring represents down-regulated genes; purple coloring represents genes from database; the shade is related to the change fold (darker colors indicate more apparent differences).
ARTICLE HIGHLIGHTS
Research background
Taurine is a beta amino acid with a simple structure and mostly appears in the free state in organism, and it plays a protective role against several forms of hepatic damage, including hepatic fibrosis. However, the molecular mechanism of taurine remains unclear, and therefore, it is difficult to use taurine for precision therapies in liver diseases.
Research motivation
The main topics of this study included investigating taurine-induced changes in gene expression in human HSCs, subjecting all of the differential expressed genes to gene ontology function and Kyoto encyclopedia of genes and genomes pathway enrichment analysis, exploring the interactions of differentially expressed genes (DEGs) in a human protein-protein interaction(PPI) network, and clarifying the mechanism of taurine on human hepatic stellate cells (HSCs).The findings enhance the understanding of the molecular mechanism of taurine-induced HSC apoptosis and provide references for liver disorder therapy.
Research objectives
The present study aimed to establish a network including transcriptomic and protein-protein interaction data, and to elucidate the molecular mechanism of taurine-induced HSC apoptosis.
Research methods
Raw microarray data were obtained as “.IDAT” and “.SDF” format using Genome Studio software (Illumina, San Diego, CA, United States), and then imported into the R environment for further processing. Subsequently, quantile normalization was carried out in R using the lumi package with Bioconductor open source software (http://www.bioconductor.org/). Significance was calculated using at-test without multiple testing correction, selecting all transcripts with a minimum change in expression level of 1.5-fold together with aP-value less than 0.05. An enrichment analysis was then conducted using the Database for Annotation, Visualization and Integrated Discovery online tool, and the PPI network was constructed using Cytoscape software. Furthermore, the MCODE plug-in of Cytoscape was used to conduct a sub-module analysis.
Research results
It was found that nine critical genes, includingMMP2,MMP9,MMP21,TIMP3,KLF10,CX3CR1,TGFB1,VEGFB, andEGF, were screened in the PPI network analysis.
Research conclusions
The present study showed thatVEGFB, EGF, BDNF, TGFB1, FIGF, TIMP3, FGB, EGFR, F7,MMP9, MMRN1, andTCOF1play key roles in the action mechanism of taurine, and these DEGs are mainly involved in positive regulation of protein kinase B signaling, ERK1 and ERK2 cascade, MAP kinase activity, MAPK cascade, cell migration, and cell proliferation.
Research perspectives
This study preliminarily explored the molecular mechanism of taurine on HSCs. Future studies should focus on the following aspects. First, the present study had no validation data and the findings could not be generalized to other conditions. As a result, future research should conduct the validate experiments, including the differentially expressed genes and the p38 MAPK-JNKCaspase9/8/3 pathway by using blockersviathe method of PCR and Western blot. Second, the characters of active and quiescent HSCs are different and the two subtypes should be studied separately in the future.
 World Journal of Gastroenterology2019年9期
World Journal of Gastroenterology2019年9期
- World Journal of Gastroenterology的其它文章
- Liver stem cells: Plasticity of the liver epithelium
- Reaction of antibodies to Campylobacter jejuni and cytolethal distending toxin B with tissues and food antigens
- Computed tomography scan imaging in diagnosing acute uncomplicated pancreatitis: Usefulness vs cost
- Targeted puncture of left branch of intrahepatic portal vein in transjugular intrahepatic portosystemic shunt to reduce hepatic encephalopathy
- Optimized protocol of multiple post-processing techniques improves diagnostic accuracy of multidetector computed tomography in assessment of small bowel obstruction compared with conventional axial and coronal reformations
- Comprehensive lifestyle intervention vs soy protein-based meal regimen in non-alcoholic steatohepatitis