
肿瘤中ERG基因的表达及其临床研究进展
2019-03-07 11:31:40王文君於宇崔久嵬
天津医药 2019年2期
王文君,於宇,崔久嵬
ERG基因(ETS-related gene)位于21号染色体q22(21q.22.2)上,属于 E26转化特异性(E26 transformation-specific,EST)转录因子家族。ERG基因参与生理性和病理性血管生成、调节血管发育、控制内皮分化和重编程、维持外周血小板数量、参与正常巨核细胞(megakaryocytes,MEG)生成等[1]。近年来越来越多的研究发现,ERG基因在一些肿瘤中过表达,在肿瘤发生发展中发挥重要作用。ERG与其他基因易位产生融合基因产物可导致ERG过表达,进而调控ERG的靶基因和下游信号通路,参与肿瘤发生发展,例如前列腺癌(prostate cancer,PCa)、尤文肉瘤(Ewing′s sarcoma,EWS)和白血病等[2]。在白血病中,ERG本身作为癌基因也可以发生过表达。但是ERG在肿瘤中的具体作用机制尚未完全明确[3]。目前,关于ERG在肿瘤中的诊断、靶向治疗和预后成为研究热点,为肿瘤治疗提供了新的方向。本文就ERG基因的结构功能、与肿瘤的关系以及靶向治疗做一综述。
1 ERG基因的结构
ERG基因跨越282个千碱基对(kilobase,kb),含有至少12个外显子和3个近端启动子(PⅠ-Ⅲ)。转录起始下游85 kb的区域已被鉴定为ERG增强子。在正常造血过程中该增强子具有活性,ERG通过其正向调节自身表达[1]。ERG的C′端有一个高度保守的ETS DNA结合结构域,可以识别含有核心GGA(A/T)的DNA序列,介导DNA结合和转录激活。N′端含有蛋白激酶C和PNT结构域的磷酸化位点,PNT结构域可形成螺旋-环-螺旋结构,供蛋白质-蛋白质相互作用和同/异二聚化[1],见图1。
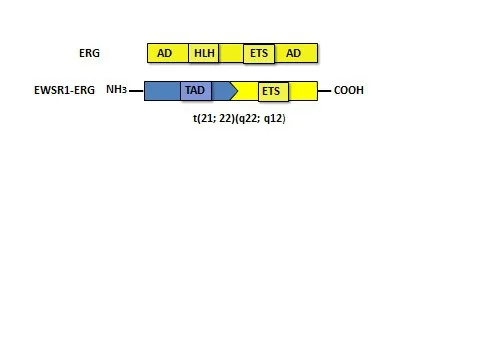
Fig.1 Diagram of translocation of ETS-related gene as well as EWSBR1 in ERG and Ewing′s sarcoma图1 ERG和尤文肉瘤中EWSBR1和ERG易位的示意图
2 ERG基因的正常表达与功能
正常情况下,ERG表达仅限于在内皮细胞(endothelial cell,EC)、MEG和软骨细胞中。在ETS家族中,ERG在EC中表达最高,几乎所有EC特异性表达的基因在其启动子中都具有ETS结合位点[1]。在胚胎发育期间和出生后,ERG在胚胎发育、血管生成、造血系统、细胞凋亡等方面发挥重要作用。
在胚胎发育方面,ERG通过结合心内膜间质转化(endocardial-mesenchymal transformation,EnMT)调控因子的启动子和内含子来上调锌指转录因子Snail家族表达,介导EnMT。EnMT减少而导致ERG突变体胚胎的心内膜垫形成缺陷,无法发育为成熟的功能瓣叶。
ERG还可促进血管成熟、稳定性和连接的完整性[1]。ERG通过调节多个EC基因的表达,如血管生成素-1、claudin-5(CLDN5)、细胞间黏附分子 2(intercellular adhesion molecule 2,ICAM-2)等[1]。在造血系统中,ERG是正常血液干细胞发育所需的ETS家族转录因子。ERG通过诱导造血干细胞(hematopoietic stem cell,HSC)自我更新因子的几种基因表达,抑制HSC中原癌基因MYC驱动的HSC分化,从而调节HSC自我更新和分化之间的平衡。ERG还参与生成正常MEG、维持外周血小板(platelet,PLT)数量。ERG属于造血干细胞和祖细胞(hematopoietic stem and progenitor cells,HSPC)表达的转录因子七肽(包括 GATA2、LYL1、TAL1、ERG、FLI1、RUNX1和LMO2)。
在成熟的脉管系统中,ERG促进稳态基因表达、抑制炎症相关基因表达,如ICAM-1、纤溶酶原激活物抑制剂等,抑制细胞因子诱导的EC激活。在细胞凋亡方面,ETS转录因子参与各种细胞类型的细胞凋亡,并且通常作为保护性因子。最后,ERG是滑膜关节形成机制的重要组成部分,可以调节大多数滑膜细胞的发育行为。
3 ERG基因在肿瘤中的表达与临床意义
ERG的表达具有组织特异性,作为谱系决定转录因子,ERG的过表达与肿瘤发病机制有关,包括促进肿瘤细胞增殖和肿瘤血管生成两方面。
在肿瘤细胞增殖方面,在细胞分裂发生染色体易位过程中,ERG与其他基因发生易位,产生融合基因产物导致ERG过表达,例如PCa中雄激素调节跨膜丝氨酸蛋白酶(transmembrane protease,serine 2,TMPRSS2)与ERG融合形成TMPRSS2-ERG融合基因,EWS中的Ewing肉瘤断裂区域1(Ewing sarcoma breakpoint region 1,EWSR1)基因和FUS RNA结合蛋白(FUS RNA binding protein,FUS)基因分别与ERG融合形成EWSR1-ERG和FUS-ERG融合基因,白血病中也形成FUS-ERG融合基因。另外,一些白血病中ERG作为癌基因发生过表达。ERG过表达后调控ERG靶基因和下游相关信号通路,发挥促肿瘤作用。
ERG还在肿瘤血管生成中发挥重要作用,血管生成是肿瘤生长和侵袭转移的重要因素。Strzepek等[4]发现,相比ERG阴性PCa,ERG阳性PCa中有更高的微血管密度,而微血管密度是许多肿瘤的预后因素。肿瘤血管生成是由于血管生成因子和抑制因子之间的失衡,ERG诱导Notch配体DLL4和其他血管生成基因表达,在促进肿瘤新血管生成方面有重要作用[1]。血管内皮生长因子(vascular endothelial growth factor,VEGF)/丝分裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)/细胞外信号调节激酶(extracellular signal-regulated kinase,ERK)信号传导通路控制ERG活性,ERG磷酸化后招募p300至VEGF依赖性增强子来调节许多血管生成基因表达,即组成VEGF/ERK/ERG/p300转录途径[5]。而靶向ERG磷酸化,选择性阻断血管生成和维持血管稳定性,可能利于降低ERG的致癌活性[5]。
3.1 ERG在PCa中的表达及临床研究进展 PCa是全球癌症死亡的第五大常见原因,发病机制尚不清楚,且治疗方法局限,且患者易发展为去势抵抗性前列腺癌,预后差[6]。2005年,Tomlins等[7]首次发现PCa中存在一种复发性基因组重排,即TMPRSS2和ERG基因之间发生融合,在高加索人、美国人中发生率为40%~60%,在激素难治性PCa中达50%~70%,在PCa的发生发展中有重要临床意义。
3.1.1 PCa中的TMPRSS2-ERG融合基因及作用机制 前列腺上皮细胞通常不表达ERG,而基因融合导致ERG成为PCa中最常见的过表达原癌基因,并且驱动前列腺上皮内瘤变向PCa转变。在PCa中,TMPRSS2的5′端非翻译区和ETS家族基因[包括ETS 变体 1(ETV1),ETV4 和 ETV5]发生融合[7-8]。ERG和TMPRSS2是21号染色体长臂上相距3 Mb的2个基因,并且ERG内含子2以及TMPRSS2内含子1和2中均存在脆弱的位点和断点。
ERG和TMPRSS2由于染色体易位或基因间片段缺失而融合形成TMPRSS2-ERG融合基因。雄激素信号募集雄激素受体(androgen receptor,AR)和拓扑异构酶Ⅱβ至TMPRSS2和ERG基因的调控区域内的断点区域,从而诱导DNA双链断裂和基因融合;因此,这种融合被认为是由长期暴露于雄激素、AR活性增强和阻止DNA双链断裂的蛋白质PIWIL1的抑制造成的[9]。此外,TMPRSS2-ERG基因融合还受ER依赖性信号调节:在高级前列腺上皮内瘤变中,ERα上调并介导雌二醇的致癌作用,在去势抵抗性前列腺癌中,ERβ作为肿瘤抑制因子发生部分缺失,从而促进TMPRSS2-ERG基因融合[10]。最新研究发现,BET溴结构域蛋白家族成员中的溴结构域蛋白4(Bromodomain containing 4,BRD4)参与修复DNA双链断裂和非同源末端连接途径,促进TMPRSS2-ERG 融合[11]。
然而TMPRSS2-ERG融合基因作用机制尚不清楚。TMPRSS2的启动子含有雄激素敏感元件,雄激素存在时,该融合基因驱动ERG过表达,调控一系列的靶基因和信号通路,从而抑制前列腺细胞分化和增加PCa细胞侵袭性,促进PCa的发生与发展。
几乎在所有PCa中,AR都高表达。一方面,AR驱动TMPRSS2-ERG基因融合;另一方面,该融合基因反之破坏AR信号传导,从而导致细胞处于去分化状态和恶性转化。此外,ERG还解离多梳抑制复合物2,生成多梳抑制复合物2亚基EZH2,上调组蛋白去乙酰化酶和致癌转录因子c-Myc,来抑制细胞分化,促进PCa的恶性转化[12-13]。
ERG过表达可抑制15-羟基前列腺素脱氢酶(15-hydroxyprostaglandin dehydrogenase,HPGD),从而阻断前列腺素E2(prostaglandin E2,PGE2)分解代谢,PGE2积聚还导致尿激酶型纤溶酶原激活物(urokinase-type plasminogen activator,uPA)活化和细胞生长,ERG还直接与uPA的启动子结合,上调PGE2受体4(E prostanoid receptor type 4,EP4);ERG可与 SPOP(Speckle-type POZ protein)同时发生突变,促进PCa侵袭和转移;ERG过表达上调基质金属蛋白酶-9(matrix metalloproteinase-9,MMP-9)和PLXNB1(plexin B1)的表达[14-15];ERG 过表达激活Notch信号,从而激活Hes(Hairy/Enhancer of split)和Hey-1(hairy/enhancer-of-split related with YRPW motif protein 1),促进PCa细胞增殖。Notch信号还可上调上皮-间质转化(epithelial-to-mesenchymal transition,EMT)转录因子;ERG过表达时有磷酸酶和张力蛋白同系物(phosphatase and tensin homologue,PTEN)失活或缺失,随后通过PTEN/AKT/PIK3/mTOR途径加速PCa的发展[16];ERG过表达导致EZH2(enhancer of Zeste Homolog 2)生成,EZH2催化组蛋白H3K9和H3K27三甲基化,招募DNA甲基转移酶(DNA methyltransferase,DNMT);ERG过表达还上调组蛋白去乙酰化酶(histone deacetylases,HDACs)表达和抑制组蛋白乙酰转移酶(Histone acetyltransferases,HATs)。ERG还可以结合其自身启动子内的ETS基序,即反式激活自身启动子,形成正反馈循环,与PCa细胞的侵袭性增加有关,见图2。

Fig.2 The role of TMPRSS2-ERG fusion gene in the pathogenesis of prostate cancer图2 TMPRSS2-ERG融合基因在PCa发病机制中的作用
3.1.2 TMPRSS2-ERG融合基因在PCa中的临床研究进展 在诊断、监测进展风险和预后方面,TMPRSS2-ERG融合基因与高侵袭性和预后不良的PCa相关,免疫组织化学染色法检测和ERG抗体可以有效诊断PCa及其转移灶[17]。TMPRSS2-ERG融合基因检测有助于优化临床风险分层、预测主动监视期间PCa的进展风险、提高预后准确性;其也是PCa患者接受前列腺切除术治疗最重要的预后因素[18]。联合分析 TMPRSS2-ERG、前列腺抗原 3(PCA3)和高级别前列腺上皮内瘤变可提高特异度和敏感度,减少前列腺组织活检[19-20]。
在治疗方面,首先,靶向TMPRSS2-ERG融合基因及相关信号通路的靶向治疗可以作为PCa治疗的新方向。目前,恩杂鲁胺(Enzalutamide)和阿比特龙均已被美国食品药品监督管理局(FDA)批准广泛应用于转移性去势抵抗性前列腺癌的治疗,这2种药物显著提高总体生存率,但很快出现耐药[21],因此需要更有效的靶向药物。靶向抑制TMPRSS2-ERG融合基因表达可引起PCa细胞生长明显停滞。BET抑制剂通过抑制BRD4表达,进而抑制DNA修复基因在前列腺细胞中的表达和非同源末端连接DNA修复途径,从而减少TMPRSS2-ERG融合[11]。目前有两项关于iBET762的临床试验正在进行中(ClinicalTrials.gov;NCT01587703,NCT01943851)。此外,ERG抑制肽和衍生肽类可以特异性结合和水解ERG蛋白,从而阻断ERG蛋白与DNA和蛋白质之间的相互作用,治疗前景广阔[22]。
其次,Notch通路抑制剂可间接抑制TMPRSS2-ERG表达,延缓TMPRSS2-ERG阳性PCa的生长,并且提高对各种肿瘤常规疗法的药物敏感性。小分子γ-分泌酶抑制剂1是Notch通路抑制剂,可以抑制TMPRSS2-ERG阳性PCa细胞的生长,并增加其对雄激素生物合成抑制剂和AR抑制剂的敏感性[15],但其作用未完全确定[23]。另外,2种HDAC抑制剂,抗癫痫药物丙戊酸/2-丙基戊酸(Valproic acid/2-propylpentanoic acid,VPA)和曲古抑菌素-A,可结合HDAC催化位点,从而抑制Ⅰ、Ⅱ类HDAC和AR的活性,并且影响p53的乙酰化状态,上调p21/WAF1/CIP1,抑制TMPRSS2-ERG的表达,从而抑制PCa的发生与发展。Notch和AR抑制剂的组合疗法、同时靶向EZH2和组蛋白去乙酰化酶的一些联合治疗均有治疗前景[15]。
3.2 ERG在EWS中的表达及临床研究进展 EWS是继骨肉瘤后第二大原发恶性骨癌,治疗主要依靠放疗、化疗和分子靶向治疗[24]。85%~90%的EWS患者中存在一种特异性的t(11;22)易位,最常见的融合基因包括 EWS-FLI1[t(11;22)(q24;q12)]、EWSBR1-ERG[t(21;22)(q22;q12)]等,还有罕见的FUS-ERG融合阳性EWS。鉴于EWS细胞形态学具有非特异性,这种基因融合可以改进EWS的鉴别、分类、诊断和分子靶向治疗[2]。
3.2.1 EWS中的EWSBR1-ERG和FUS-ERG融合基因及作用机制 RNA结合TET家族的N′端转录激活结构域可以和ETS家族的C′端DNA结合结构域发生重排。在典型EWS中,TET家族(包括EWSR1或FUS)与ETS家族(包括FLI1、ERG、ETV1、ETV4或FEV)的基因之间发生融合。最常见的融合基因是EWS-FLI1,其次是22q12上EWSR1基因与ETS家族成员之间发生融合,如ERG、ETV1、ETV4和FEV[2]。致癌融合蛋白仅在结合RNA解旋酶A之后才能具有活性。
EWSR1融合蛋白的表达干扰内源性EWSR1功能,而内源性EWSR1在促进DNA修复因子聚集到DNA损伤部位中起关键作用,并且通过改变选择性剪接来降低几种DNA损伤应答基因的表达水平。因此,EWSR1融合蛋白导致EWS中的基因组不稳定性和高水平复制应激(replication stress,RS),从而导致内源性DNA损伤[25]。携带致癌基因诱导的RS的EWS细胞依赖于共济失调-毛细血管扩张有关激酶(Ataxia telangiectasi and Rad3 related,ATR)/细胞周期检测点激酶1(Checkpoint kinase 1,CHK1)存活,在EWS中观察到ATR和CHEK1过度表达和活化,CHK1通过下调RS和防止ATM/半胱天冬酶-3依赖性细胞死亡,从而增加EWS细胞的增殖和生存能力[26]。其次,ERG等异常的转录因子反式激活其他基因,参与EWS的发生与发展。如具有DNA修复、细胞分化、增殖和肿瘤转化功能的多聚腺苷二磷酸核糖聚合酶[poly(ADP-ribose)polymerase 1,PARP-1],其启动子中转录起始位点周围存在ETS结合位点,EWSBR1-ERG和FUS-ERG融合产物驱动PARP1过表达,进而促进转录激活,形成正反馈环,与EWS细胞的增殖相关。此外,EWSBR1-ERG融合蛋白激活转录抑制因子HDAC和其他阻遏物,来抑制视黄醇类 X 受体 α(Retinoid X receptor α,RXRα)及其靶基因的正常转录活性。RXRs作为一种核受体与靶基因启动子中的特定序列结合而起转录因子作用,可以介导类视黄醇包括天然维甲酸及其合成衍生物的抗癌功能。
3.2.2 EWSR1-ERG和FUS-ERG融合基因在EWS中的临床研究进展 在诊断方面,荧光原位杂交技术检测EWSR1突变是诊断EWS的金标准,但存在假阴性。因此在具有典型形态学和(或)强CD99和ERG免疫反应的情况下,应用一些分子测试可提高复杂基因重排的诊断率,例如荧光原位杂交技术或实时逆转录-聚合酶链反应检测[27]。
在治疗方面,多种靶向药物试验正在进行中。PARP抑制剂奥拉帕尼可通过抑制PARP1修复EWS细胞的DNA,减少EWS细胞的分裂和侵袭,但少数ETS结构域内存在点突变时无效。一项前瞻性Ⅱ期临床试验(ClinicalTrials.gov;NCT01583543)中,奥拉帕尼在标准化疗失败后的晚期EWS患者中安全且耐受性良好。目前,关于奥拉帕尼和替莫唑胺、伊立替康等化疗药物联合应用的临床试验正在积极进行中(ClinicalTrials.gov;NCT01858168)。此外,CHK1/ATR抑制剂作为单一药物通过多种机制增强RS,从而导致DNA损伤积累和随后的细胞死亡,因此可作为潜在疗法[25]。再者,干扰致癌融合蛋白和RNA解旋酶A相互作用的新型生物靶向小分子,例如YK-4-279,可抑制ERG和ETV1依赖的转录活性,从而降低EWS细胞的运动性和侵袭性[28]。最后,VPA作为HDAC抑制剂,可以抑制异常融合蛋白EWS-ERG和EWS-FLI1在EWS细胞中的表达,从而增加EWS细胞对细胞凋亡的易感性,并且减轻EWSBR1-ERG融合蛋白对RXRα转录活性的抑制,恢复RXRα靶基因RARβ、CRABP2和p21的抑癌活性,因此VPA具有治疗潜力,但尚未见相关临床试验。
3.3 ERG在白血病中的表达及临床研究进展 研究发现,在一些白血病中也出现ERG过表达,并且ERG高表达与不同类型血液恶性肿瘤的总体生存率和无病生存率相关。此外,ERG高表达还可以作为正常细胞遗传学的急性髓系白血病(acute myeloid leukemia,AML)、T细胞急性淋巴细胞白血病(T-acute lymphoblastic leukemia,T-ALL)和儿童急性淋巴细胞白血病(Acute lymphoblastic leukemia,ALL)等独立的预后不良因素[29-31]。
3.3.1 白血病中的ERG基因及作用机制
3.3.1.1 T-ALL 在早期T淋巴细胞生成过程中ERG表达出现下调,但在T-ALL中ERG正常表达。正常T细胞中ERG+85 kb增强子无活性,而在TALL中该增强子高度活跃,且针对造血干细胞和胸腺T细胞祖细胞,导致ERG表达。其他T细胞癌基因SCL/TAL1,LMO2以及ETS(ERG,FLI1)等转录因子,与ERG+85 kb的增强子结合,介导ERG高表达。
此外,ERG阳性白血病中出现几种癌症相关信号通路上调,其中MAPK/ERK信号通路最为明显。MAPK/ERK信号传导介导ERG3 S283的高度磷酸化,因此在白血病细胞中ERG3磷酸化的丝氨酸283(phosphorylation of serine 283,pS283)高于正常HSPC,并导致与MAPK/ERK活性相关的基因激活,pS283 ERG突变蛋白在ERG+85 kb增强子处富集导致内源性ERG转录增加,以及促进CD34+HSPC增殖[32]。
3.3.1.2 AML 研究表明,有复杂核型和21号染色体异常的AML患者均有ERG转录水平的上调[33]。Salek-Ardakani等[34]认为ERG作为一种癌基因可以促进各种谱系成熟造血细胞的生长,对髓系白血病发病机制和维持至关重要。与T-ALL一样,在AML中MAPK/ERK信号通路也发挥同样作用,导致ERG的过表达。
ERG的靶基因包括GATA2和HEY2等转录因子,GATA基序在ERG结合的区域内高度富集,从而导致GATA2和HEY2特异性地在ERG阳性的髓系白血病中过表达。HEY2和GATA2参与早期造血功能,GATA2是造血干细胞和MEG发育的关键调节因子,高GATA2表达是儿童AML不良预后因素;而HEY2被认为是心血管发育的调节因子,参与白血病的发生与进展。ERG过表达还诱导具有高度耐药表型的间充质样状态,从而使肿瘤细胞获得生长优势[35]。
此外,有研究认为ERG通过结合PIM1的启动子和增强子来诱导PIM1激酶癌基因表达。PIM激酶可以磷酸化组蛋白H3、c-Myc、Mcl-1、p27、Bad和FLT3,从而增加了c-Myc驱动的转录水平、抑制细胞凋亡、促进细胞增殖、增加FLT3-ITD介导的耐药性,涉及癌症进展和对化疗药物的耐药性,高表达ERG和PIM1的AML预后较差[36]。
3.3.1.3 急性巨核细胞白血病(acute megakaryoblastic leukemia,AMKL) ERG还和AMKL有关,已发现定位于21号染色体的唐氏综合征关键区域的ERG与唐氏综合征相关的巨核细胞白血病有关。ERG是MEG生成的正调控因子,可结合SCL+19增强子,调节造血祖细胞中SCL/TAL1的表达,而SCL1/TAL1过表达会使祖细胞朝向MEG谱系分化。已知GATA1也是调控MEG生成中重要的转录因子,几乎存在于所有唐氏综合征相关AMKL患者。ERG和GATA1可影响信号通路PI3K/AKT/mTOR。活化的AKT可以与ERG、GATA1协同促进MEG异常增殖。
3.3.1.4 白血病中的FUS-ERG融合基因 除了EWS,在一些慢性粒细胞白血病急变期、急性白血病、AML 和ALL 中,也发现染色体易位 t(16;21)(p11;q22)导致了FUS与ERG融合,破坏天然的FUS RNA结合结构域,插入ERG DNA结合结构域。FUS基因位于16p11,编码转录激活因子,通过结合RNA polⅡ和一些转录因子参与转录起始,从而产生异位转录激活。FUS外显子1~6,7或8以及ERG外显子的9,10或11到C′末端参与融合基因形成。FUS-ERG不仅影响骨髓谱系,还影响淋巴谱系或更原始的造血细胞。这种白血病抵抗化疗且复发率高,预后很差。此外,FUS-ERG融合蛋白具有双重转化活性,取决于其N′末端结构。FUS-ERG融合蛋白还干扰RNA的剪接。FUS的N′末端可以结合RXR,因此FUS-ERG融合蛋白也是视黄酸信号传导途径的转录阻遏物。
3.3.2 ERG及其融合基因在白血病中的临床研究进展 在诊断和预后方面,由于白血病皮肤和皮肤中反应性粒细胞浸润在病理学上难以区分,而ERG可以作为用来区分的免疫组织化学标志物来区分两者,Xu等[37]在白血病患者皮肤活检中检测到ERG阳性率高达81.4%(13/16),特异度为100%。另外,在ALL中,ERG与ALL中的糖皮质激素(GCs)耐药性有关,ERG在GCs敏感细胞中被招募到GCs受体的启动子上,这是GCs依赖性细胞凋亡所需要的,而在耐药细胞中未观察到这种招募,并且ERG和激活蛋白-1可以作为ALL中GCs应答的决定因素[38]。
在治疗方面,PIM抑制剂SGI-1776对FLT3和PIM激酶具有双重抑制作用,能够降低组蛋白H3、c-Myc、Mcl-1、p27、Bad和FLT3的磷酸化,可以抑制ERG阳性的白血病细胞生长并诱导其凋亡,抑制效果与PIM1的表达水平相关[36]。但由于SGI-1776有心脏剂量限制性毒性,其临床试验已撤回(ClinicalTrials.gov;NCT01239108)。用小干扰RNA敲除PIM1也可以抑制ERG阳性白血病细胞的生长。此外,RAS途径激活是ERG驱动白血病的间接靶点,PIM抑制剂和RAS抑制剂组合可增加抑制效果[36]。但在治疗过程中,ERG过表达细胞会通过与基质细胞相互作用来增加细胞存活率,还可通过抑制蛋白激酶C或Wnt信号传导途径介导凋亡逃避,从而导致耐药性。
鉴于ERG影响AKT并具有协同作用,AKT变构抑制剂MK-2206可以通过抑制AKT磷酸化来抑制PI3K/Akt/mTOR通路激活,诱导白血病细胞中G1期停滞和细胞凋亡。MK-2206还通过抑制糖原合酶激酶-3β磷酸化,导致糖原合酶激酶-3β介导的蛋白酶体依赖性抗凋亡促存活蛋白髓样细胞白血病-1(myeloid cell leukemia-1,Mcl-1;Bcl-2蛋白质家族的成员)降解,从而降低白血病细胞活力、诱导细胞凋亡。并且MK2206能够提高AML中阿糖胞苷活性。MK-2206已在复发或难治性AML患者中进行临床试验(ClinicalTrials.gov;NCT01253447)。
3.4 ERG在其他肿瘤中的表达以及临床研究进展 ERG在其他肿瘤中也有一定作用。ERG在具有软骨分化的骨和软组织肿瘤中稳定表达,可作为软骨分化的标志物。ERG是血管内皮以及血管结构衍生肿瘤高度敏感性和特异性的标志。ERG在所有血管瘤和淋巴管瘤、一些高度血管化的中枢肿瘤的内皮细胞中稳定表达[3],可作为高度特异性的良恶性血管性肿瘤的新标志物[47],但尚不清楚其是否有致癌作用。此外,ERG仅在中枢神经系统肿瘤的内皮细胞中表达,可作为特异性的中枢神经系统肿瘤内皮细胞标志物。ERG在上述三类肿瘤中研究较少,有待进一步探究。
4 展望
在多种肿瘤中ERG基因被发现过表达,成为目前肿瘤研究热点。近年来,ERG在一些肿瘤中的发现以及分子机制方面的研究逐步深入,ERG本身作为癌基因或者通过融合基因在多种肿瘤中发生过表达,从而调控下游靶基因和信号通路。尽管ERG作用机制尚未完全清楚,但给肿瘤发病机制研究提供了新思路和方向。
目前,ERG已在一些肿瘤中初步显示出治疗潜力和预后价值,成为抗肿瘤治疗中有前景的靶点和预后标志物。目前已有靶向治疗药物应用于临床,还有一些正处于临床试验阶段。但是由于ERG在正常组织中也存在表达,如何开发特异性的肿瘤靶向药物及其相应的疗效评估有待研究。在预后方面,ERG在肿瘤中可以用于监视肿瘤进展风险和提高预后准确性,显示出一定的预后价值,但还需更多前瞻性临床试验来验证。此外,鉴于ERG基因在肿瘤血管生成方面的作用,ERG在EC中高表达提示肿瘤微血管丰富,因此靶向ERG抑制肿瘤血管生成和ERG作为潜在的预后标志物均具有研究前景。综上所述,ERG基因在部分肿瘤的发病机制、治疗、预后等方面显现出初步的研究价值,为肿瘤的发病机制研究、精准治疗和预后评估提供了新的方向。
猜你喜欢
军事文摘(2024年2期)2024-01-10 01:59:00
保健医苑(2022年5期)2022-06-10 07:46:38
中国临床医学影像杂志(2021年6期)2021-08-14 02:21:56
肝博士(2020年5期)2021-01-18 02:50:18
河南畜牧兽医(2017年12期)2017-11-13 04:05:18
上海农业学报(2017年3期)2017-04-10 12:39:26
中国科技信息(2015年6期)2015-11-10 03:35:44
医学研究杂志(2015年7期)2015-06-22 11:01:01
中国当代医药(2015年16期)2015-03-01 02:03:13
中国药理学通报(2014年2期)2014-05-09 08:22:39
