
超声辅助合成纳米氧化锰及其低浓度NO去除性能研究
2019-03-07 06:43龚云刘艳顾萍朱钰方周晓霞
无机材料学报 2019年2期
龚云, 刘艳, 顾萍, 朱钰方, 周晓霞
超声辅助合成纳米氧化锰及其低浓度NO去除性能研究
龚云1,2, 刘艳3, 顾萍1, 朱钰方2, 周晓霞3
(1. 上海出版印刷高等专科学校 印刷包装工程系, 上海 200093; 2. 上海理工大学 材料科学与工程学院, 上海 200093; 3. 中国科学院 上海硅酸盐研究所, 上海 200050)
通过超声辅助的工艺, 利用氧化还原法制备出多价态纳米MnO。通过调节超声时间、KMnO4浓度、烘干温度、反应溶液pH, 探索MnO的最佳合成条件。结果表明: 超声时间20 min, 高锰酸钾浓度0.5 mol/L, 烘干温度80 ℃, 反应溶液pH=7的条件下, 合成的样品MnO表现出最佳的催化性能, 对100%的NO去除率可持续15 h。采用X射线衍射分析(XRD)、N2-吸脱附测试、扫描电镜(SEM)、透射电镜(TEM)等技术考察最优催化剂MnO的结构和形貌, 借助X射线光电子能谱(XPS)和傅里叶红外分析(FT-IR)研究最优催化剂MnO对NO去除的催化氧化机理以及催化剂的失活机制。结果表明, 三维贯穿的多级孔结构, 花瓣状的形貌和弱晶化的晶体结构有利于气体吸附和传输。多价态Mn和氧空位的存在促进了NO和O2的吸附和激活, 因此最优样品MnO表现出优异的NO的常温催化氧化。
MnO; 低浓度NO; 超声; 纳米材料; 氧化
城市半封闭空间虽然缓解了汽车拥堵和停车难等顽症, 但是机动车冷启动或频繁启动时尾气温度常低于催化剂有效发挥作用的最低温度, 受到自然通风的限制, 这些尾气无法及时排出, 导致地下空间的空气质量严重恶化。根据文献[1-2]报道, 其主要污染物氮氧化物(NO, 其中NO占90%以上)的浓度高达数ppm甚至数十ppm (1 ppm= 0.001‰)。因此, 开发常温治理技术来解决半封闭空间中低浓度NO的污染显得非常紧迫和必要。传统的NO治理技术有选择性催化还原法[3-4](Selective Catalytic Reduction, SCR)、选择性非催化还原法[5-6](Selective Non-Catalytic Reduction, SNCR)和存储-催化还原法(NOStorage-Reduction, NSR)[7-8], 工作温度均在200 ℃以上, 不能满足常温NO治理的需要。除此之外, 固相吸附法[9]和液相吸收法[10]可以在常温下进行低浓度NO2的治理, 然而半封闭空间中的NO主要是NO, 而低水溶性的NO很难在常温下被吸附或吸收。因此, 在常温下将NO氧化成化学活性高、水溶性好的NO2, 是实施吸附/吸收法的必要前提。由此可见, 构建高效的催化剂开展常温低浓度NO的催化氧化具有重要的意义。
目前, 用于NO常温催化氧化的材料主要包括活性炭、分子筛以及过渡金属氧化物。Isao Mochida等[11]发现活性炭纤维对浓度为380 cm3/m3的NO具有良好的常温催化氧化效果, 其催化氧化活性位点可能与活性炭纤维在热处理过程中伴随CO/CO2化合物的释放所诱导的不饱和碳有关。刘华彦等[12-13]报道离子改性的高硅ZSM-5分子筛对浓度500 cm3/m3的NO具有较好的常温催化氧化活性, 这是因为异质原子掺杂在沸石中诱导了大量的酸性活性位和氧空位, 促进NO和O2的吸附, 生成吸附态的NO3, 并继续与NO作用生成弱吸附的NO2和N2O4, 吸附饱和后释放出来[12]。沸石分子筛与活性炭的催化过程类似, 在反应初始发生NO的吸附, 当吸附达到饱和后, NO被氧化成NO2。除此之外过渡金属氧化物具有丰富的电子结构和价态, 它们及其复合氧化物被相继合成用于NO的催化氧化, 尤其是锰基氧化物。Huang等[14]通过共沉淀法制备了一系列Mn系的复合氧化物, 如Fe-Mn、Fe-Mn-Cu、Fe-Mn-Co、Fe-Mn-Ni、Fe-Mn-Ce、Fe-Mn-Zr、Fe-Mn-Ti等, 研究发现它们对500 cm3/m3的NO具有良好的常温催化氧化及吸附活性, 可在反应初期长时间实现100% NO的去除。上述催化剂对高浓度的NO具有良好的催化活性, 但是当NO浓度降低后其催化性能骤然下降。这是因为在动力学上, NO被O2氧化的速度与NO的浓度平方成正比, 其反应速度随NO初始浓度减小而急剧降低。因此, 必须探索其他催化剂用于低浓度NO的常温催化去除。
本课题组前期在Mn系氧化物的合成以及高效低浓度NO常温去除方面取得了一系列的进展[15-18]。本工作在此基础上, 利用超声辅助的氧化还原法, 通过调节超声时间、反应物浓度、烘干温度、反应溶液pH制备弱晶化的多价态MnO系列样品, 研究其在常温下对低浓度NO的去除性能。在最优工艺制备MnO催化剂的基础上, 对催化剂进行一系列物化性能表征, 研究其对低浓度NO的常温催化氧化机理以及催化剂失活机制。
1 实验方法
1.1 催化剂的制备
高锰酸钾(KMnO4, 分析纯, 国药集团化学试剂有限公司), 去离子水, 无水乙醇(C2H6O, 优级纯, 国药集团化学试剂有限公司)。
通过KMnO4和乙醇的氧化还原反应制备弱晶化的MnO, 继而调节超声时间、反应物的浓度、反应溶液pH和烘干温度, 考察不同条件下合成的弱晶化MnO对NO催化氧化性能的影响。所得样品标记为MnO-, 其中表示超声时间, 单位是min;表示反应浓度, 单位为mol/L;表示烘干温度, 单位为℃;表示反应溶液pH。具体实验步骤如下:
(1)调节超声时间: 1.58 g KMnO4溶于20 mL H2O, 逐滴加入到10 mL无水乙醇中, 滴加结束后置于超声池中超声0、20、40、60 min。用去离子水洗涤过滤干净, 置于80 ℃烘箱烘干14 h。所得样品标记为MnO-0, MnO-20, MnO-40, MnO-60。
(2)调节反应浓度: 分别制备0.1、0.5、1.0 mol/L KMnO4(20 mL H2O)的水溶液, 逐滴加入到10 mL无水乙醇中, 滴加结束后置于超声池中超声20 min, 用去离子水洗涤过滤干净, 在80 ℃烘箱中烘干14 h。所得样品标记为MnO-20-0.1, MnO-20-0.5, MnO-20-1。
(3)调节烘干温度: 1.58 g KMnO4溶于20 mL H2O, 逐滴加入到10 mL无水乙醇中, 滴加结束后置于超声池中超声20 min, 用去离子水洗涤过滤干净, 分别在80、120、160 ℃烘箱中烘干14 h。所得样品标记为MnO-20-0.5-80、MnO-20-0.5-120和MnO- 20-0.5-160。
(4)调节反应溶液pH: 1.58 g KMnO4溶于20 mL H2O, 逐滴加入到10 mL无水乙醇中, 分别滴加0.5 mL 的HNO3(1mol/L), 0.5 mL的H2O, 0.5 mL的KOH (1mol/L)。超声20 min后, 用去离子水抽滤, 洗涤, 烘干温度为80 ℃, 烘干时间为14 h。所得样品标记为MnO-20-0.5-80-4, MnO-20-0.5-80-7和MnO-20-0.5-80-9。
1.2 催化剂的表征
在Rigaku D/MAX- 2200PC型X射线衍射仪上进行样品的X射线衍射(XRD)分析, Cu靶, Ka射线, 管路电压40 kV, 管电流40 mA, 扫描速度为4°/min, 室温进行, 扫描范围2= 10°~80°。在Micromeritics TriStar 3020型号孔径分析仪上测试样品的N2吸附脱附曲线, 先将样品放在N2气中进行150 ℃预处理6 h, 再在液氮温度77 K下进行N2吸附-脱附实验。根据Brunauer-Emmett-Teller (BET)算法得到材料比表面积数值, Barrett-Joyner-Halenda (BJH)方法模拟得到材料孔径分布图。在Magellan 400型场发射扫描电镜和JEOL 200CX型场发射透射电镜上观察样品形貌, 透射电镜加速电压为200 kV。在Thermo Scientific ESCALAB 250型X射线光电子能谱仪上进行XPS分析, 以AlKa为激发光源, 入射光子能量1486.6 eV, 激发功率15 kW, 用C的1s电子结合能285 eV进行误差校正。在Nicolet iS10型傅里叶红外分析仪上进行FT-IR分析, 分辨率为4 cm–1。
1.3 NO去除催化性能评价
0.1 g的催化剂装载在内径为8 mm的石英管固定床中, 反应温度通过水浴控制在25 ℃。原料气N2、O2和NO经质量流量计控制配合后, 调控NO浓度为10 cm3/m3、O2含量为21%、N2为载气, 反应气流量200 mL×min–1, 空速为120 000 mL×g–1×h–1。进口及出口气体中的NO与NO2浓度通过NO分析仪(Thermo Fisher 42i-LS)在线监测。NO去除率通过以下公式计算可得:
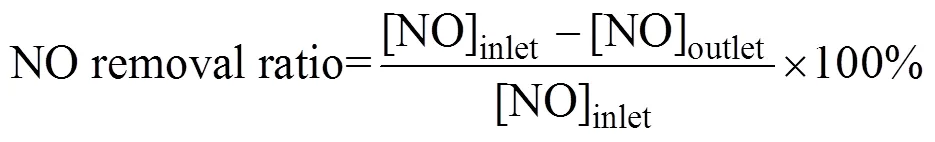
这里以催化剂MnO完全去除NO以及去除80% NO所持续的时间作为评价催化活性的指标。
2 结果和讨论
2.1 MnOx对NO去除性能的影响因素
在催化剂的制备过程中, 分别调节超声时间、反应物浓度、烘干温度以及反应溶液pH, 以期获得具有最佳NO催化去除性能的合成工艺。
超声处理可以在材料中制造缺陷, 从而提高活性位点覆盖率, 促进催化反应的进行[18]。因此, 可以通过调节超声时间来研究材料MnO对NO去除性能的影响。研究表明: 未超声样品(MnO-0)和超声时间过长的样品(MnO-60)对NO的去除效果都很差, 而超声时间为20和40 min的样品(MnO-20和MnO-40), 对NO的100%去除效果能维持10 h以上, 如图1(a)所示。这是由于超声时间过长会造成MnO结构坍塌, 从而覆盖部分活性位点。而只有合适的超声时间, 才能使MnO暴露更多的活性位点, 从而使得催化剂具有最优的催化活性, 因此最优的超声时间为20 min。
反应物浓度是影响材料结构和性能的重要原因之一, 因此可以通过改变KMnO4的初始浓度来调控反应浓度, 研究其对NO去除性能的影响, 结果如图1(b)所示。KMnO4初始浓度为0.5 mol/L, 制备的样品MnO-20-0.5表现出最优的催化活性, NO的100%去除率可以维持6 h, 而KMnO4初始浓度为0.1和1 mol/L时, MnO-20-0.1样品几乎没有催化活性, MnO-20-1样品能够维持约4 h的100% NO去除率。这是由于反应物浓度过低, 成核速率较缓慢, 晶粒就生长较快, 导致产物结构相对致密并且缺陷较少; 而反应物浓度过高, 反应成核速率过快, 晶粒生长缓慢, 导致颗粒尺寸不均匀, 容易形成无定形的结构, 影响材料的稳定性, 导致催化剂失活。
除此之外, 实验发现烘干温度对于催化剂MnO性能的影响也很大, 如图1(c)所示。当烘干温度为80 ℃时, 样品MnO-20-0.5-80的100% NO去除率可以维持10 h以上。随着烘干温度升高, 样品对NO去除性能显著降低,尤其是当烘干温度为120和160 ℃时, MnO-20-0.5-120和MnO-20-0.5-160样品的性能近乎直线下降, 这可能是因为过高的烘干温度会影响样品MnO表面吸附水和结合水的含量和比例, 从而影响NO的氧化性能。
进一步地, 本课题组也探索了溶液pH对催化剂性能的影响。如图1(d)所示, 当溶液pH为7时, 样品MnO-20-0.5-80-7的100% NO去除率可持续15 h, 大于80%的NO去除率可持续18 h, 具有最优的催化性能。而溶液pH为4和7得到的样品, 对NO的去除率无法达到100%且催化时间持续不长。由此可见, 溶液pH对样品MnO的催化活性有着显著的影响, 这可能是由于酸碱环境容易破坏氧化锰的结构。

图1 MnOx样品合成工艺参数对NO催化氧化性能的影响
(a) Ultrasonic time; (b) Reactants concentration; (c) Dry temperature; (d) pH
综上所述, 通过调节超声时间、反应物浓度、烘干温度和反应溶液pH, 探索出超声辅助醇水溶液法制备MnO的最佳合成条件为超声20 min, 高锰酸钾浓度0.5 mol/L, 烘干温度80 ℃, 反应溶液pH=7, 在此条件下合成的样品MnO-20-0.5-80-7, 其100% NO去除率可持续15 h, 大于80%的NO去除率可持续18 h, 具有最佳的NO催化性能。对该最优样品进行后续的表征和性能测试, 研究其反应机理和失活机制。
2.2 XRD物相分析
图2(a)是样品MnO-20-0.5-80-7在400 ℃煅烧前后的XRD图谱。未煅烧的样品呈弱晶化态, 峰形不明显。400 ℃煅烧后样品的XRD图谱中出现MnO2的衍射峰。与标准卡片对比, 发现制备的MnO-20- 0.5-80-7结构属于四方晶系(I4/m(87))[19]。除此之外, MnO-20-0.5-80-7的衍射峰包含两个较宽的峰, 对应的层间距为0.24 nm (2=37°)和0.14 nm (2= 67°), 分别对应(211)晶面和(112)晶面。XRD图谱表明经过超声辅助工艺制备的弱晶化MnO-20-0.5-80-7主要选择暴露(211)和(112)晶面。
2.3 N2吸脱附结构性能分析
图2(b)是样品MnO-20-0.5-80-7的N2吸附-脱附曲线和相应的孔径分布曲线, 从图中可以发现, MnO-20-0.5-80-7属于典型的Ⅲ型曲线, 介孔主要是来自颗粒之间的堆积孔。经过计算可得, 最优的MnO比表面积为66 m2/g, 孔容高达10.3 cm3/g。通过孔径分布曲线可以发现材料具有较宽的孔径分布40~ 120 nm。样品结构中三维贯穿堆积孔的存在不仅有利于气体传输和吸附, 而且可以充分利用材料表面和内部暴露的活性位点, 从而加速催化反应的进行。

图2 样品MnOx-20-0.5-80-7煅烧前后的XRD图谱(a)和N2吸附-脱附曲线与孔径分布曲线(b)
2.4 形貌分析
图3(a)~(b)是样品的低倍和高倍SEM照片, 从图中可以看出合成的MnO-20-0.5-80-7呈现纳米花的形貌。其中, 敞开的片状纳米花瓣可以为催化反应提供更多的反应场所, 从而有利于NO的吸附和催化氧化。从TEM照片(图 3(c)~(d))也可以看出MnO-20-0.5-80-7的片状形貌结构。另外, 高倍TEM照片对应的电子衍射环很弱, 表明样品主要呈现无定形的结构。然而, 样品部分区域仍有一些晶格条纹(红色图框所示), 表明样品呈弱晶化状, 与XRD结果一致。这种弱晶化的MnO-20-0.5-80-7相对晶化度高的样品具有更多的缺陷, 可以为催化反应提供更多的活性位点, 从而提高催化活性。
2.5 XPS表面化学成分分析
为了表征MnO-20-0.5-80-7催化反应前后表面的化学元素组成和元素价态分布, 对样品进行XPS分析, 结果如图4(a)~(d)所示。对Mn2p3/2、O1s轨道进行分峰拟合, 相应结果如表1所示。其中, 未经刻蚀处理的样品表征的主要是样品表面的性质, 刻蚀处理过的样品表征的是样品近体相的性质。MnO-20-0.5-80-7可以在641.2、642.5和643.5 eV拟合为三个峰, 分别对应于Mn2+、Mn3+、Mn4+[16-17]其中Mn4+含量最高, 占66.5%。经过催化性能测试后Mn3+增多(37.6%), Mn4+(58.3%)减少。经过刻蚀过的样品, 主要以Mn3+的形式存在, 而且经过催化反应之后, Mn2+、Mn3+和Mn4+的含量基本变化不大。
以上的结果说明MnO-20-0.5-80-7对于NO的催化去除主要源于Mn3+和Mn4+之间的氧化还原转化, 而刻蚀后的催化剂表面Mn价态变化不大, 所以NO的催化氧化主要发生在材料的表面。

图3 MnOx-20-0.5-80-7的SEM (a)~(b)和TEM (c)照片以及相应的电子衍射图(d)

图4 催化反应前后MnOx-20-0.5-80-7的XPS谱图
(a) Mn2p3/2, before; (b) Mn2p3/2, after; (c) O1s, before; (d) O1s, after

表1 MnOx-20-0.5-80-7在NO催化反应前后XPS数据
O1s的XPS图谱经过分峰处理后可得到两个主峰: 530.1和531.5 eV, 如图4(c)~(d)所示, 前者可归于晶格氧(Olat), 而后者归属于吸附氧(Oads)[17]。从表1可以发现, 经过刻蚀处理的样品具有更高的表面吸附氧, 说明MnO-20-0.5-80-7材料体系中存在大量的氧空位, 有助于O2的吸附。催化反应后, 材料表面吸附氧Oads减少, 晶格氧Olat增多。而刻蚀后样品中吸附氧Oads增加、晶格氧Olat减少。表面吸附氧Oads与晶格氧Olat占比的变化表明氧物种参与了催化反应。

图5 催化剂MnOx-20-0.5-80-7对低浓度NO常温去除性能评价(进口气体: [NO]=10 cm3/m3, [O2]=21%, 载气N2, 25 ℃, 空速120000 mL×h–1×g–1)
2.6 NO催化去除性能分析
图5给出了样品MnO-20-0.5-80-7对于低浓度NO的催化去除性能, 可以发现, 样品对低浓度NO 具有良好的去除效果, 大于80%的NO去除率可以维持18 h, 而100%的NO去除率可以维持15 h。在线尾气监测显示, 整个测试过程经历了吸附、氧化、脱附三个阶段。在反应初始阶段, 在出口气体中检测不到任何NO的存在, 表明MnO-20-0.5-80-7已经完全吸附NO。随着反应的进行, 出口的NO2浓度持续增加, 而出口仍检测不到NO, 说明MnO- 20-0.5-80-7可以完全吸附NO并将其催化氧化为NO2。最后, 在出口中可以同时检测到NO和NO2, 而且NO浓度随着时间延长而逐渐增加, 而NO2浓度随时间的增加有所下降, 表明MnO-20-0.5-80-7对NO的吸附能力和催化能力都在下降, 催化剂逐渐失活, 这可能与活性位点逐渐被覆盖有关。
2.7 FT-IR分析
为了探索催化剂逐渐失活的原因, 对催化反应前后的MnO-20-0.5-80-7样品进行FT-IR测试, 结果如图6所示。相比催化反应前的样品MnO-20-0.5- 80-7, 催化反应后的样品在1050、1270、1390 cm–1附近出峰, 根据文献[20-21], 1390 cm–1隶属于硝酸盐基团峰, 1270 cm–1属于亚硝酸盐峰, 1050 cm–1属于硝酸盐对称振动峰。可见NO催化氧化后在样品表面形成了亚硝酸盐和硝酸盐, 这可能是活性位点被覆盖后样品失活的主要原因。

图6 MnOx-20-0.5-80-7催化反应前后的FT-IR谱图
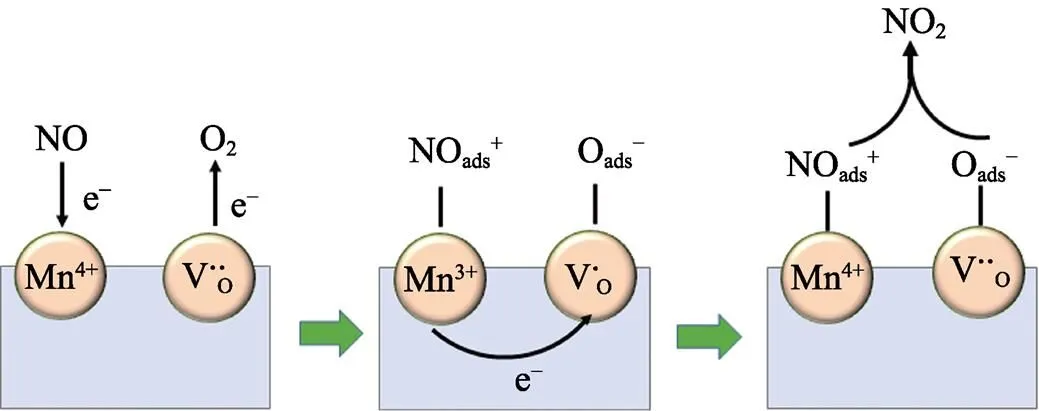
图7 在材料MnOx可能发生的NO催化氧化机理
2.8 NO催化机理示意
基于上述结果, 本研究提出了一种可能的NO催化氧化机理, 如图7所示。XPS数据中表面吸附氧比例要高于刻蚀后样品的吸附氧, 说明MnO-20- 0.5-80-7材料体系中存在一定的氧空位, 促进O2的吸附。在NO的常温催化氧化反应中, NO和O2首先吸附在Mn4+和氧空位形成NOads和Oads; 然后, Mn4+得到电子变为Mn3+同时吸附的NOads和Oads被变价为Mn3+和氧空位激活形成NOads+和Oads–,最后, 被活化的NOads+和Oads–相互反应生成NO2, 伴随着Mn3+和氧空位之间的电荷转移, Mn3+由于失去电子恢复为Mn4+。本课题组认为在整个催化反应中Mn4+和Mn3+之间的价态转化以及氧空位的参与使得NO能够被氧化成NO2, 因此多价态Mn和氧空位的存在有助于NO的常温催化氧化。
3 结论
调控不同的合成工艺参数, 得到多价态MnO的最佳合成条件: 超声时间20 min, 高锰酸钾浓度0.5 mol/L, 烘干温度80 ℃, 反应溶液pH=7。在最优条件下合成的多价态纳米MnO具有三维贯穿的多级孔结构, 花瓣状的形貌和弱晶化的晶体结构, 这种独特的孔道结构和形貌有利于气质传输和吸附。在NO的室温催化氧化反应中, 最优的样品MnO对100%的NO去除率可持续15 h, 大于80%的NO去除率可持续18 h, 表现出最优的催化氧化性能。其优异的催化性能可以归因于Mn4+和Mn3+之间的转化以及大量氧空位的存在, 促进了NO和O2的吸附和激活。最后, 通过FT-IR证实催化反应后, 最优样品表面形成了亚硝酸盐和硝酸盐, 导致了活性位点的覆盖和催化剂的失活。
[1] LMEIDA-SILVA M, CANHA N, FREITAS M C,. Air pollution at an urban traffic tunnel in Lisbon, Portugal: an INAA study., 2011, 69(11): 1586–1591.
[2] GUERRINI G L. Photocatalytic performances in a city tunnel in Rome: NOmonitoring results., 2012, 27(1): 165–175.
[3] MONTICELLI O, LOENDERS R, JACOBS P A,. NOremoval from exhaust gas from lean burn internal combustion engines through adsorption on FAU type zeolites cation exchanged with alkali metals and alkaline earth metals., 1999, 21(3): 215–220.
[4] LIU Z M, WOO S I. Recent advances in catalytic DeNOscience and technology.,2006, 48(1): 43–89.
[5] HAN X H, WEI X L, SCHNELL U,. Detailed modeling of hybrid reburn/SNCR processes for NOreduction in coal-fired furnaces.e, 2003, 132(3): 374–386.
[6] BAE S W, ROH S A, KIM S D. NO removal by reducing agents and additives in the selective non-catalytic reduction (SNCR) process., 2006, 65(1): 170–175.
[7] TAKAHASHI N, YAMAZAKI K, SOBUKAWA H,. The low-temperature performance of NO(x)storage and reduction catalyst., 2007, 70(1–4): 198–204.
[8] KLEIN J, WU D L, TSCHAMBER V,. Carbon-NSR catalyst interaction: impact on catalyst structure and NOstorage efficiency., 2013, 132: 527–534.
[9] CHANG X F, LU G Z, GUO Y,. A high effective adsorbent of NO: preparation, characterization and performance of Ca-beta zeolites., 2013, 165: 113–120.
[10] WEI J C, YU P, CAI B,. Absorption of NO in aqueous NaClO2/ Na2CO3solutions.logy, 2009, 32(1): 114–119.
[11] MOCHIDA I, KISAMORI S, HIRONAKA M,. Oxidation of NO into NO2over active-carbon fibers., 1994, 8(6): 1341–1344.
[12] LIU H Y, ZHANG Z K, XU Y Y,. Adsorption-oxidation reaction mechanism of NO on Na-ZSM-5 molecular sieves with a high Si/Al ratio at ambient temperature., 2010, 31(10): 1233–1241.
[13] LIU H Y, LI Y F, XU Y Y,. Adsorption and catalysis on the surface of high silica ZSM-5 molecular sieve in NO oxidation at ambient temperature., 2011, 25(4): 615–621.
[14] HUANG H Y, YANG R T. Removal of NO by reversible adsorption on Fe-Mn based transition metal oxides., 2001, 17(16): 4997–5003.
[15] SHU Z, CHEN Y, HUANG W M,. Room-temperature catalytic removal of low-concentration NO over mesoporous Fe-Mn binary oxide synthesized using a template-free approach., 2013, 140: 42–50.
[16] DU Y Y, HUA Z L, HUANG W M,. Mesostructured amorphous manganese oxides: facile synthesis and highly durable elimination of low-concentration NO at room temperature in air., 2015, 51(27): 5887–5889.
[17] WANG J, ZHU J Z, ZHOU X X,. Nanoflower-like weak crystallization manganese oxide for efficient removal of low-concentration NO at room temperature., 2015, 3(14): 7631–7638.
[18] DU Y Y, HUANG W M, HUA Z L,. A facile ultrasonic process for the preparation of Co3O4nanoflowers for room-temperature removal of low-concentration NO.. 2014, 57(2): 73–77.
[19] NIAG E C, CHEN C H, GENUINO H,. Total oxidation of CO at ambient temperature using copper manganese oxide catalysts prepared by a redox method., 2010, 99(1/2): 103–110.
[20] HADYIIVANOV K I. Identification of neutral and charged NOsurface species by IR spectroscopy., 2000, 42(1/2): 71–144.
[21] BENTRUP U, BRUCKNER A, RICHTER M,. NOadsorption on MnO2/NaY composite: anFT-IR and EPR study., 2001, 32(4): 229–241.
Synthesis of Nano Manganese Oxide with Assistance of Ultrasonic for Removal of Low Concentration NO
GONG Yun1,2, LIU Yan3, GU Ping1, ZHU Yu-Fang2, ZHOU Xiao-Xia3
(1. Department of Printing and Packing, Shanghai Publishing and Printing College, Shanghai 200093, China; 2. School of Material Science and Engineering, University of Shanghai for Science and Technology, Shanghai 200093, China; 3. Shanghai Institute of Ceramics, Chinese Academy of Sciences, Shanghai 200050, China)
The varied-valence nanocatalyst MnOwas prepared using the oxidation-reduction method under ultrasonic. The optimal synthetic conditions of MnOwere explored by adjusting the ultrasonic time, concentration of reaction precursor, drying temperature, and pH of reaction solution. The results indicated that the optimal sample MnOwas synthesized under the condition of ultrasonic time of 20 min, 0.5 mol/L KMnO4, drying temperature of 80 ℃ and pH=7, which showed the super catalytic performance and the time of 100% NO removal rate was as high as 15 h at room temperature. The structure and morphology of the optimal catalyst MnOwere investigated by XRD, N2-adsorption-desorption analysis, SEM and TEM. Besides, XPS and FT-IR were also applied to explore the catalytic oxidation process of NO removal and the deactivation mechanism of the optimal sample MnO. It is believed that the interpenetrating and hierarchal pore, the petaloid morphology and weak crystallization structure contribute to the gas adsorption and transmission. The presence of varied-valence Mn and oxygen vacancy can improve the adsorption and activation of NO and O2, thus, enhancing the NO catalytic removal on the optimal catalyst MnOat room temperature.
MnO; low concentration NO; ultrasonic; nano materials; oxidation
TQ174
A
1000-324X(2019)02-0186-07
10.15541/jim20180209
2018-05-04;
2018-08-14
柔版印刷绿色制版与标准化实验室开放课题基金(2BKT201803) Lab of Green Platemaking and Standardization for Flexographic Printing (2BKT201803)
龚云(1987–), 女, 讲师. E-mail: 331391649@ qq.com
周晓霞, 助理研究员. E-mail: zhouxiaoxia@mail.sic.ac.cn
猜你喜欢
中国资源综合利用(2022年9期)2022-10-13
科学导报(2022年32期)2022-06-09
绿色科技(2022年8期)2022-05-25
现代矿业(2022年3期)2022-04-09
保鲜与加工(2021年1期)2021-02-06
中国科技纵横(2017年7期)2017-05-16
江苏农业科学(2017年1期)2017-02-27
爆笑show(2015年3期)2015-05-08
妇女生活(2014年11期)2014-09-10
中国高新技术企业(2014年7期)2014-06-06
