
Discovery of leaf region and time point related modules and genes in maize (Zea mays L.) leaves by Weighted Gene Co-expression Network analysis (WGCNA) of gene expression pro files of carbon metabolism
2019-02-14 03:12WANGJingluZHANGYingPANXiaodiDUJianjunMALimingGUOXinyu
WANG Jing-lu, ZHANG Ying, PAN Xiao-di, DU Jian-jun, MA Li-ming, GUO Xin-yu
Beijing Key Lab of Digital Plant/Beijing Research Center for Information Technology in Agriculture/National Engineering Research Center for Information Technology in Agriculture/Beijing Academy of Agriculture and Forestry Sciences, Beijing 100097, P.R.China
Abstract Maize (Zea mays L.) yield depends not only on the conversion and accumulation of carbohydrates in kernels, but also on the supply of carbohydrates by leaves. However, the carbon metabolism process in leaves can vary across different leaf regions and during the day and night. Hence, we used Weighted Gene Co-expression Network analysis (WGCNA) with the gene expression pro files of carbon metabolism to identify the modules and genes that may associate with particular regions in a leaf and time of day. There were a total of 45 samples of maize leaves that were taken from three different regions of a growing maize leaf at five time points. Robust Multi-array Average analysis was used to pre-process the raw data of GSE85963 (accession number), and quality control of data was based on Pearson correlation coef ficients. We obtained eight co-expression network modules. The modules with the highest significance of association with LeafRegion and TimePoint were selected. Functional enrichment and gene-gene interaction analyses were conducted to acquire the hub genes and pathways in these significant modules. These results can support the findings of similar studies by providing evidence of potential module genes and enriched pathways associated with leaf development in maize.
Keywords: WGCNA, maize leaf, gene expression, gene modules, pathways
1. lntroduction
Maize (Zea maysL.) is the largest food crop in the world and one of the five staple crops in China. It is important for food security, and thus is a crucial part of agricultural production.In recent years, consumer demand for maize has been increasing. Consequently, improving the yield of maize has become one of the most important research topics in crop science. It is well known that plant yield primarily depends on the accumulation and distribution of photosynthetic assimilates. Leaves, the photosynthetic “source” organs,and, specifically, carbon metabolism in leaves are keys to improve maize yield.
Carbon metabolism, also known as carbohydrate metabolism, is a crucial process in plant metabolism.Driven by photosynthesis, the assimilation of carbon results in the synthesis of carbohydrate products. The form of carbohydrate can be divided into two types: non-structural and structural. Non-structural carbohydrates, such as sucrose, starch, and soluble sugar, mainly participate in the metabolic processes of plant life. Structural carbohydrates,such as cellulose and lignin, are primary components of the morphological structure of the plant. Starch, which is stored in leaves, is the main form of leaf photosynthetic products.At night, the starch is hydrolyzed by amylase to produce sucrose. Subsequently, sucrose is transported through the phloem to the leaves. Generally, a high carbohydrate content in a “source” organ with a large capacity for storing sucrose is bene ficial to the growth and development of maize.
Many studies are available in literatures on plant growth and carbon metabolism in plants (Ben-Haj-Salah and Tardieu 1995; Wieseet al. 2007; Poireet al. 2010;Gonzalezet al. 2012; Apeltet al. 2015; Avramovaet al.2015; Czedik-Eysenberget al. 2016). Czedik-Eysenberget al. (2016) examined carbon metabolism in maize, by defining a developmental gradient to aid the analysis of carbon metabolism in the meristem, elongation, and mature regions of maize leaves. They used the metabolome and transcriptome and polyribosome loading reactions as qualitative indicators of protein synthesis, and tracked the elongation of the entire leaf at five different time points.A series of analyses was then used to identify candidate genes by searching for differentially expressed transcripts associated with carbon metabolism between the growth and mature regions. The gene expression data generated in their study were deposited in the Gene Expression Omnibus(GEO, http://www.ncbi.nlm.nih.gov/geo/) and has been invaluable to peer research including our own. Because their original analyses did not analyze all the chip probe sets, we endeavored to further analyze the data by using Weighted Gene Co-expression Network analysis (WGCNA).Gene modules that significantly relate to certain traits (such as LeafRegion and TimePoint) could then be found.
WGCNA is a method in systematic biology used to describe gene-related patterns in microarray samples(Langfelder and Horvath 2008). It has been successfully used in a variety of biological contexts and is becoming increasingly popular in bioinformatics applications. Given the large amount of microarray data, WGCNA is a valuable tool for excavating and interpreting the multiple forms of expression of genes in different biological samples.WGCNA can be used to find the significant modules that are associated with different traits. Overall, WGCNA is a powerful data mining tool of differentially expressed genes with multiple experimental designs.
In this study, we conducted a WGCNA of gene expression data sets in relation to diurnal cycle and carbon starvation in gradients (cell division, elongation, and mature zones) of growing maize leaves. After pre-processing of raw data and performing quality control on the gene expression data, we obtained 42 maize leaf samples. All of the probe sets in the array platform were used to complete the WGCNA. Finally,the probe sets were divided into eight co-expression network modules represented by eight different colours. Some of these modules showed significant associations with specific traits. In order to explore the biological significance and functions of the genes, pathway enrichment and gene-gene interaction analyses were also carried out for the genes in each module.
2. Materials and methods
2.1. Data pre-processing and quality control
The data set used in this study was downloaded from the GEO database (Barrettet al. 2011). The GEO database includes both raw and pre-processed data. Depending on the research purposes of various researchers who submitted data, the pre-processed methods applied on the same data set may not be consistent. Therefore, we downloaded the raw data of GEO accession number GSE85963 for our study.The platform number for data acquisition is GPL4032, and the chip type is [Maize] Affymetrix Maize Genome Array.There were a total of 45 samples of maize leaves that were taken from three different regions (cell division, elongation,and mature zones) of a growing maize leaf at five time points(dawn, dusk, and 6, 14 and 24 hours after dusk). The ‘scan date’ stored in the CEL files were used as batch information,and chips run on different days were regarded as different batches (Mistryet al. 2013).
The main processes of chip data pre-processing includes data filtering, standardization, and logarithmization. Robust Multi-array Average (RMA) (Bolstadet al. 2003; Irizarryet al.2003) is a normalization approach that use median polish to summarize the perfect matches, and it can take advantage of Affymetrix data to generate an expression matrix. Hence,we chose RMA as the pre-processing method since it enables both data normalization and logarithmization. The“rma” function of R packageaffy(Gautieret al. 2004) was used to complete this procedure. For quality control, outlier samples were identi fied by pair-wise Pearson correlations.We also drew a sample dendrogram as an auxiliary means of quality control. Those samples showing correlations of less than 0.8 with all the other samples were regarded as outlier samples.
2.2. WGCNA
We used the Statistical Software R, version 3.4.1 (R Core Team 2017), for all WGCNA computations (Zhang and Horvath 2005; Langfelder and Horvath 2008) to explore the co-expression network of all 17 734 probe sets. The number of probe sets analyzed in our study is different from the original study of GSE85963, which only used the probe sets that significantly matched to a single maize gene model and retained only one probe set per gene model (Czedik-Eysenberget al. 2016). In this study, the gene annotation was completed after the gene modules were clustered. In addition, when there were multiple probe sets corresponding to the same gene in the detected gene module, we regarded the gene as a significant result with greater credibility than the other genes in this module.
The soft threshold power was calculated by the pickSoftThreshold function. The most suitable power was selected to construct a network based on the criterion of approximate scale-free topology. As shown in Fig. 1,when the power value is 6, the scale-free topology fit index curve flattened out upon reaching a high value. Hence,we chose 6 as the power to use in subsequent analysis.We set the maxBlockSize to 20000 to accommodate our large number of probe sets. In order to find the best enrichment results, multiple parameter combinations were used to compute the significant modules. Finally, the“blockwiseModules” function was used to construct the gene network, and the parameter settings were as follows:power=6, TOMType=“unsigned”, maxBlockSize=20 000,minModuleSize=200, mergeCutHeight=0.2, verbose=3. The other parameters were set to default values. The module eigengenes were subsequently calculated for each module.In addition, each module was colour-coded and assigned a network before visualization of detected modules.
The correlation between modules and traits was analyzed by the “cor” function in the R package stats (R Core Team 2017). In addition, the “corPvalueStudent” function was used to calculate the student asymptoticP-value for each correlationviathe R package WGCNA (Langfelder and Horvath 2008). The result ofP-value<0.05 was considered to be a significant correlation between the module and the trait.
2.3. Gene annotation
The latest maize gene chip annotation file “Maize Annotations, CSV format, Release 35 (4.8 MB, 10/7/14)” downloaded from the affymetrix website (http://www.affymetrix.com/support/technical/byproduct.affx?product=maize) was used in this study. The annotation file contains about 14 101 gene symbols, 14 100 Entrez IDs, and 11 600 Ensembl annotation results. In general, nearly 80% of the probe sets in the[Maize] Affymetrix Maize Genome Array are annotated.
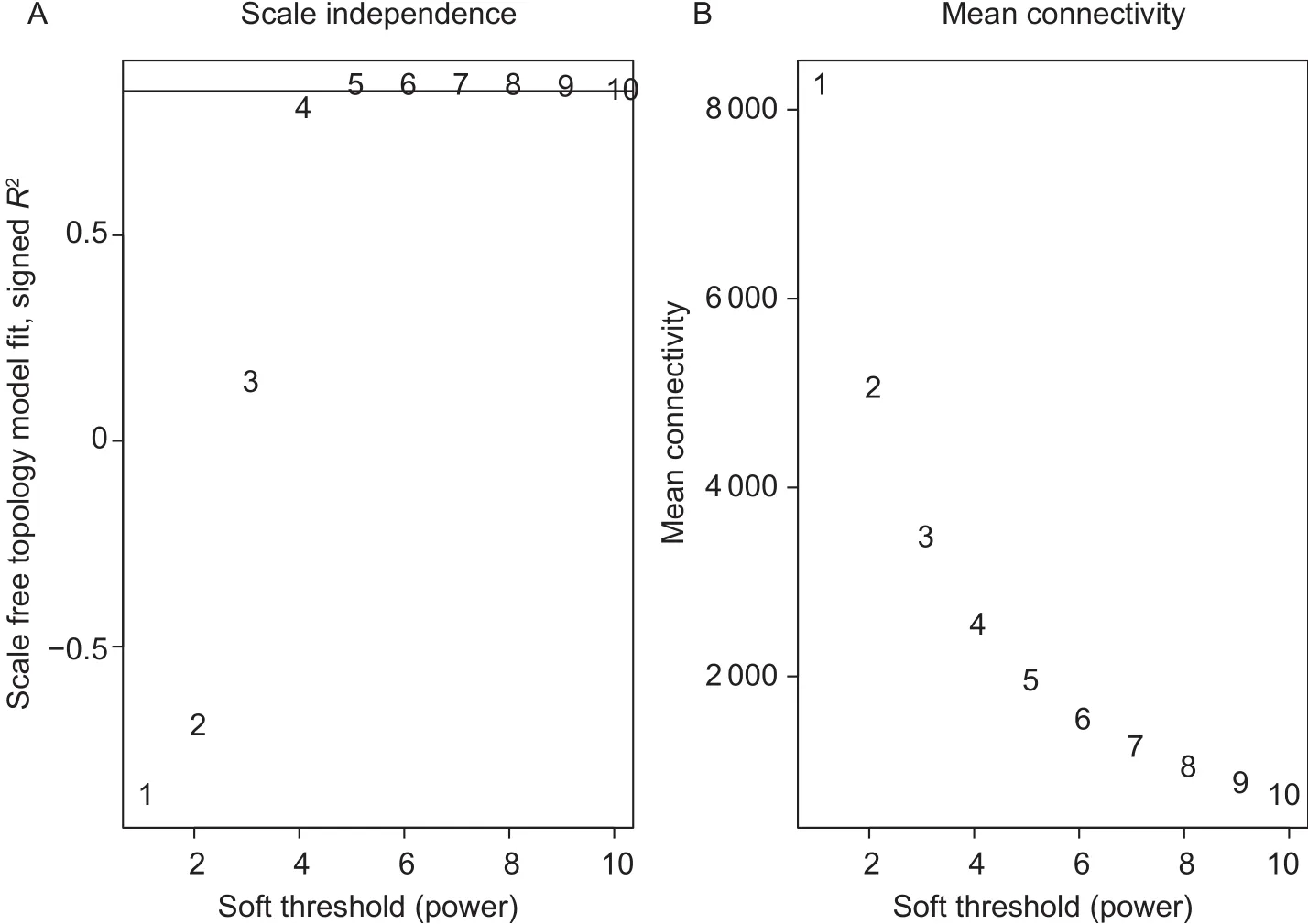
Fig. 1 The decision of power value. A, the horizontal axis represents different power values. The vertical axis represents the scale-free fit index as a function of the soft-thresholding power. The line represents that the correlation coef ficient is equal to 0.85.B, the average network connectivity under different power values.
In our study, the probe sets in each colour module were annotated, and the genes corresponding to the significant probe sets were considered as significant results. However,because several genes were mapped by multiple probe sets, we only reported the number of unique genes found within each module.
2.4. Functional analysis
DAVID 6.8 (Huanget al. 2009a, b) was applied to perform the enrichment analysis of the modules based on the genes in each module. The list of of ficial gene symbols of the genes in each module were uploaded to DAVID individually,and the default options were used to conduct the functional annotation. Pathways from the Gene Ontology (GO)(Ashburneret al. 2000) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa 2002) were used and those with aP-value<0.05 were considered as significant results.
2.5. Gene-gene interaction network analysis
In this study, we used two ways to explore the gene-gene interactions of the genes in each module. First, the original WGCNA output of this study was divided into separate files,one for each module, which contained data on gene-gene correlations with transcription factors. Then, the gene-gene interactions were visualized by Cytoscape v3.5.1.
To further evaluate the presence of biological interactions between the genes of each module, we constructed a new gene-gene interaction network among the module genes by using the maize gene-gene interaction network data from an online protein-protein interaction database for maize (PPIM)(Zhuet al. 2016) (http://comp-sysbio.org/ppim). The genegene interactions between the genes in each module and other genes were also individually visualized by Cytoscape v3.5.1. The hub genes (genes located in the center of the network) in each network were reported as the main results.
3. Results
3.1. Samples after quality control
Three samples (GSM2288631, GSM2288632, and GSM2288633) were removed based on a combination of results from Pearson correlation coef ficients and the sample dendrogram. These outliers were consistent with the original study (Czedik-Eysenberget al. 2016). Therefore,these three samples were excluded before subsequent analysis. The final sample size was 42. The final samples were individually obtained from the cell division, elongation and mature zones of maize leaf at five different time points(dawn, dusk, and 6, 14, and 24 hours after dusk). The details of the data sets are shown in Appendix A. The dendrogram of the 42 samples used for our analysis with sample information is shown in Appendix B.
3.2. Modules and eigen-genes
By using the parameters and functions in the method of WGCNA, all modules on each branch below“mergeCutHeight” were merged. Subsequently, the 17 734 probe sets were divided into eight modules where each colour in Fig. 2 represents one unique gene module.The numbers of probe sets in each module are 10 288,2 467, 1 372, 881, 856, 622, 460, and 437 for the turquoise,blue, brown, yellow, green, red, black, and pink colours,respectively (Table 1). In addition, 351 probe sets that did not merge to any module were grouped into the grey module.The system clustering tree generated by the dynamic hybrid algorithm based on dissTOM is shown in Fig. 2.
After gene annotation of each module, the probes were annotated by corresponding unique gene symbols. When more than one probe corresponded to the same gene,we retained that gene once and regarded it as a result with greater credibility than other genes. The numbers of unique genes in each module were 7 036, 2 157, 1 187,787, 777, 573, 397, and 403 for the turquoise, blue, brown,yellow, green, red, black, and pink colours, respectively.The detailed gene information of each module are given in Table 1.

Fig. 2 Identifying result of the system clustering tree by dynamic hybrid algorithm based on dissTOM. The top figure represents the system clustering tree based on dissTOM. The bottom figure of Tree Cut Merged and Merged Dynamic represent network modules before and after merging modules (each color represents one module).
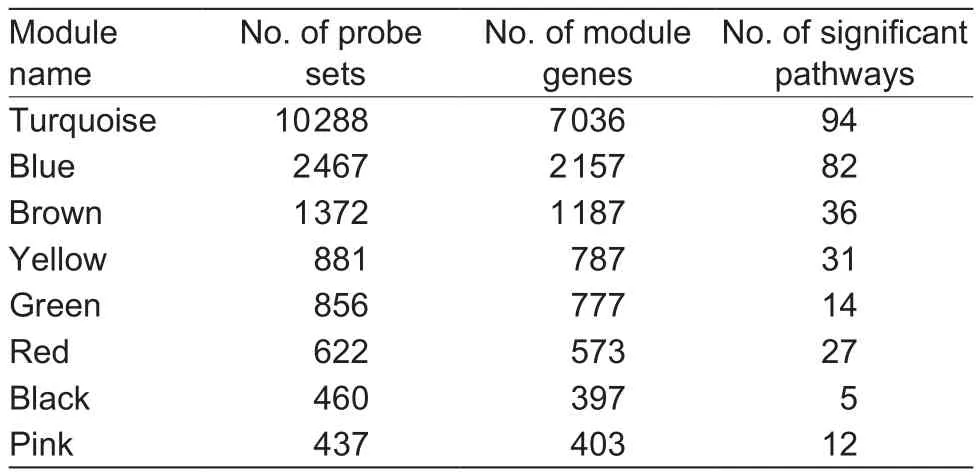
Table 1 Detailed results of each module
The correlation between the modules and the traits,LeafRegion and TimePoint, were analyzed and data are shown in Fig. 3. Four modules significantly associated with LeafRegion: Blue (P-value=9.09E–21, cor=–0.9433),Red (P-value=8.99E–10, cor=–0.7829), Turquoise(P-value=2.69E–08, cor=0.7366), and Black (P-value=3.22E–07, cor=–0.6953). The modules that highly correlated to TimePoint were Green (P-value=3.86E-13,cor=0.8580) and Pink (P-value=8.99E-10, cor=–0.7829).Additionally, Brown had significant correlation with both LeafRegion (P-value=2.14E-05, cor=0.6056) and TimePoint(P-value=1.10E-05, cor=0.6220).
3.3. Pathways of each module
We performed separate pathway enrichment analyses for the genes in each module to explore their related functions and obtained many significant KEGG pathways and GO terms for each module. Among them, the most striking pathways with strong association to LeafRegion or TimePoint are shown in Table 2. The findings of the functional analyses for each module are described below.
The most significant module associated with LeafRegion was the Blue module. A total of 82 significant pathways(P-value<0.05) were obtained after the enrichment analysis of 2 157 genes in this module. Among them, the pathway with the highest significance contained the GO term “DNA replication” (GO:0006260,P-value=3.60E-12)followed by GO terms, “DNA repair” (GO:0006281,P-value=3.00E-07) and “DNA recombination” (GO:0006310,P-value=1.30E-05). The KEGG pathways with significant associations with LeafRegion included “Spliceosome”(zma03040,P-value=2.40E-08), “RNA transport” (zma03013,P-value=7.20E-08), and “DNA replication” (zma03030,P-value=3.30E-07). The details of the enrichment pathways of Blue module are shown in Appendix C. In addition, a total of 27 significant pathways (P-value<0.05) were obtained after the enrichment analysis of 573 genes in the Red module. Ninety-four significant pathways were obtained in the Turquoise module, which contained 7 036 unique genes.Five significant pathways were obtained after the enrichment analysis of 397 genes in the Black module. The specific pathway names and the corresponding statistical results of the modules are shown in Appendices D–F.

Fig. 3 Correlation between each module and the traits(LeafRegion and TimePoint). Numbers in rectangular represent the correlation coef ficients and numbers in brackets indicate the corresponding P-value.
The most significant module related to TimePoint was the Green module. A total of 14 significant pathways(P-value<0.05) were obtained after the enrichment analysis of 777 genes in this module. Among them, the representative pathways with high significance of the KEGG pathways were: “protein processing in endoplasmic reticulum” (zma04141,P-value=5.10E-03), “valine, leucine and isoleucine degradation” (zma00280,P-value=8.40E-03),“peroxisome” (zma04146,P-value=0.023), and“benzoxazinoid biosynthesis” (zma00402,P-value=0.026);and of the GO terms were: “translation initiation factor activity” (GO:0003743,P-value=2.90E-03) and “4 iron, 4 sulfur cluster binding” (GO:0051539,P-value=6.60E-03).The details of the enrichment pathways of Green module are shown in Appendix G. Additionally, 12 significant pathways(P-value<0.05) were obtained after the enrichment analysis of 403 genes in the Pink module. The specific pathway names and their statistical results are shown in Appendix H.
The Brown module significantly associated with both LeafRegion and TimePoint. The 36 pathways were obtained after the enrichment analysis of 1 187 genes in this module.The typical KEGG pathways of Brown module were: “carbon metabolism” (zma01200,P-value=4.30E-10), “citrate cycle”(TCA cycle) (zma00020,P-value=7.90E-09), “biosynthesis of antibiotics” (zma01130,P-value=2.30E-07), “biosynthesis of amino acids” (zma01230,P-value=8.40E-07), “ubiquitin mediated proteolysis” (zma04120,P-value=6.70E-06),and “glycolysis/gluconeogenesis” (zma00010,P-value=2.90E-04). The common GO term was “tricarboxylic acid cycle” (GO:0006099,P-value=9.00E-06). The other pathways of this module can be found in Appendix I. In addition, the pathways enriched by the genes in Yellow module are shown in Appendix J.
3.4. Networks and hub genes
The original WGCNA output of this study was divided into files containing data on gene-gene correlations with transcription factors for each module. Then, the gene-gene interactions were visualized by Cytoscape v3.5.1. Because the relationship between genes is complex, there were many nodes in the network output by WGCNA, too many to warrant further mining of gene functions by WGCNA. Therefore, we used the PPIM database to further excavate the gene-gene relationship of genes in each module. Finally, we obtained the total network diagrams formed by the module genes and their related genes in each module, individually. And the networks only composed by module genes were shown in Fig. 4, which could more clearly demonstrate the results of network analysis.
The network analysis results of the modules significantly related to LeafRegion. In the total network of Blue module,there were 12 727 edges and 2 499 nodes, and 786 of the nodes were the genes of Blue module. GRMZM2G033626(also known asLOC103649975, 26S proteasome non-ATPase regulatory subunit 14 homolog), which has 341 related genes, was the hub in the total network. And in the network, only composed by Blue module genes,GRMZM2G033626 was also the hub gene with 52 related genes (Fig. 4-A). The whole network of Red module included 2 422 edges and 961 nodes, and 214 of the nodes were the genes of Red module. The hub of this network was GRMZM2G047855 (also known ascka2, CK2 protein kinase alpha 2) with 111 edges. And GRMZM2G047855 was as well as the hub gene in the network only composed by Red module genes (Fig. 4-D). In the total network of Turquoise module, there were 34 523 edges and 5 595 nodes, and 2 235 of the nodes were the genes of this module. The hub was GRMZM2G419891 (also known asMubC5, polyubiquitin) with 1 020 related genes. However,the hub gene with 54 edges in the network only composed by Turquoise module genes was GRMZM2G145258 (also known as LOC100273118, 40S ribosomal protein S3a-2).And GRMZM2G419891 was the third hub gene with 50 edges in this network (Fig. 4-B). For Black module, 1 242 edges and 550 nodes formed the total network that were obtained by enrichment analysis of genes. Moreover, 101 of the nodes were the genes of Black module. Among them,GRMZM2G068479 (also known asLOC100282531, premRNA-splicing factor ATP-dependent RNA helicase) with 83 edges was the hub gene in the whole network. No network containing only genes within Black module was generated.
The network analysis results of the modules were significantly related to TimePoint. In the total network of Green module, there were 5 277 edges and 1 860 nodes in the network, and 298 of the nodes were the genes of Green module. GRMZM2G000052 (also known asLOC101027113, lysine-specific histone demethylase 1),with 455 related genes, was the hub of this network. And it is also the hub gene of the network only formed by the Green module genes (Fig. 4-E). The total network of Pink module included 2 258 edges and 883 nodes, and 139 of the nodes were the genes of Pink module. The hub gene was GRMZM2G002100 (also known asMPK7, ABA stimulation MAP kinase). But the hub gene of the network only consisted of the Pink module genes was GRMZM2G044306(Fig. 4-G). And GRMZM2G002100 was the second hub gene in this network.
Of the Brown module, there were 7 575 edges and 2 220 nodes in the network, and 424 of the nodes were the genes of this module. In this network, GRMZM2G118637(also known aspco110026, putative ubiquitin family protein) was the hub, which related to 588 interactions.And GRMZM2G118637 with 41 edges was also the hub in the network only composed by Brown module genes(Fig. 4-C).
4. Discussion
Microarray technology provides a powerful tool for studying differentially expressed genes. Examining the differences in gene expression in different maize leaf regions and growth stages can help us understand the genes that play a role in the growth of plants. Microarray studies can provide empirical data on carbohydrate metabolism to further understanding the carbon assimilation process.Here, we report the results of a WGCNA of GSE85963 in order to improve upon past research on gene expression in carbohydrate metabolism of maize. Ultimately, eight coexpression network modules were obtained and visualized.Among them, four of the modules were highly associated with LeafRegion, and two modules were primarily related to TimePoint. The KEGG pathways and GO terms obtained by Pathway Enrichment analysis can help us understand the gene functions of each module. Furthermore, the networks generated by Gene-gene Interaction analysis can help re fine our exploration of the biological significance of each module.
In our study, several module hub genes were obtained.The gene that located at the network hub exhibited the strongest correlation with the corresponding traits. For example, GRMZM2G033626 had a high correlation with LeafRegion and, therefore, it was the hub gene of the network that was generated by Blue module genes.GRMZM2G033626, also known as LOC103649975, is the 26S proteasome non-ATPase regulatory subunit 14 homolog. It is a protein coding gene reported by various studies on maize (Gardineret al. 2004; Alexandrovet al.2009; Soderlundet al. 2009; Vega-Arreguinet al. 2009).In our analysis, GRMZM2G033626 appeared in the KEGG pathway “Proteasome” (zma03050) and was statistically significant (P-value=2.00E-04). GRMZM2G047855,the hub gene of Red module, is worthy of note as well.GRMZM2G047855 (cka2) is the protein coding gene ofCK2protein kinase alpha 2. Many studies have reported their findings on this gene (Peracchiaet al. 1999; Setny and Geller 2005; Raafet al. 2008; Lebskaet al. 2009, 2010). Lebskaet al. (2009)reported that ZmCK2alpha-4 may function as a negative regulator of otherCK2s, and under certain circumstances, is a holoenzyme whose catalytic activity is stimulated by specific regulatory subunit(s). Moreover, they subsequently found that casein kinase 2 phosphorylates the maize eukaryotic translation initiation factor 5A (eIF5A)(Lebskaet al. 2010). Raafet al. (2008) suggested that the catalytic subunit of human protein kinaseCK2structurally deviated from its maize homologue in a complex with the nucleotide competitive inhibitor emodin. GRMZM2G047855 can be found in the KEGG pathways “Circadian rhythmplant, conserved biosystem” and “Ribosome biogenesis in eukaryotes, conserved biosystem”. The hub gene of Turquoise module, GRMZM2G419891, is also a significant gene that associated with LeafRegion. GRMZM2G419891 is also known asMubC5, and it is a protein coding gene of polyubiquitin. This gene has been reported in a study on heat-shocked maize seedlings (Liuet al. 1995) and another on the maize endosperm transcriptome (Laiet al. 2004).
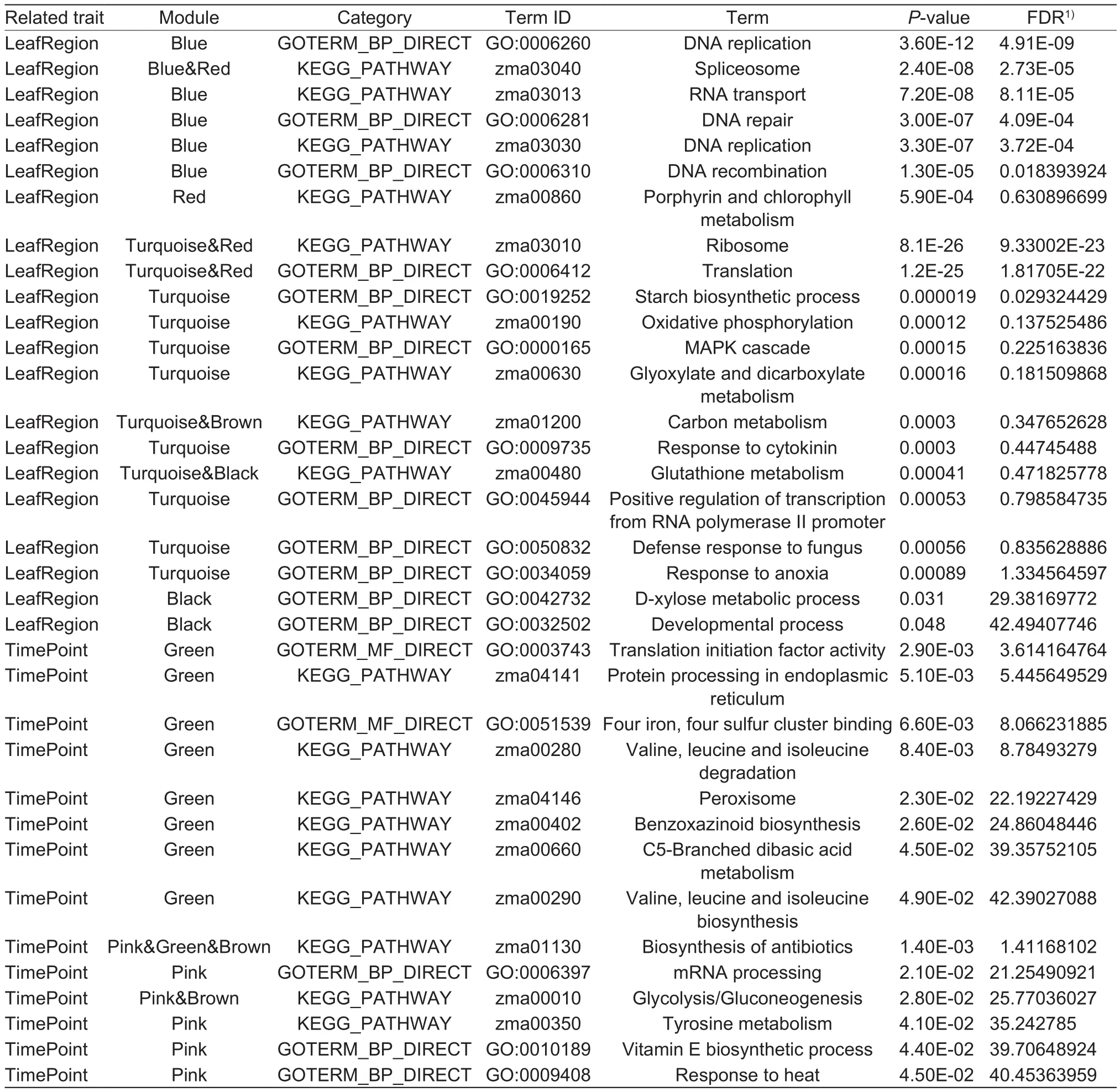
Table 2 The most striking pathways with high relevance to LeafRegion or TimePoint obtained by each module
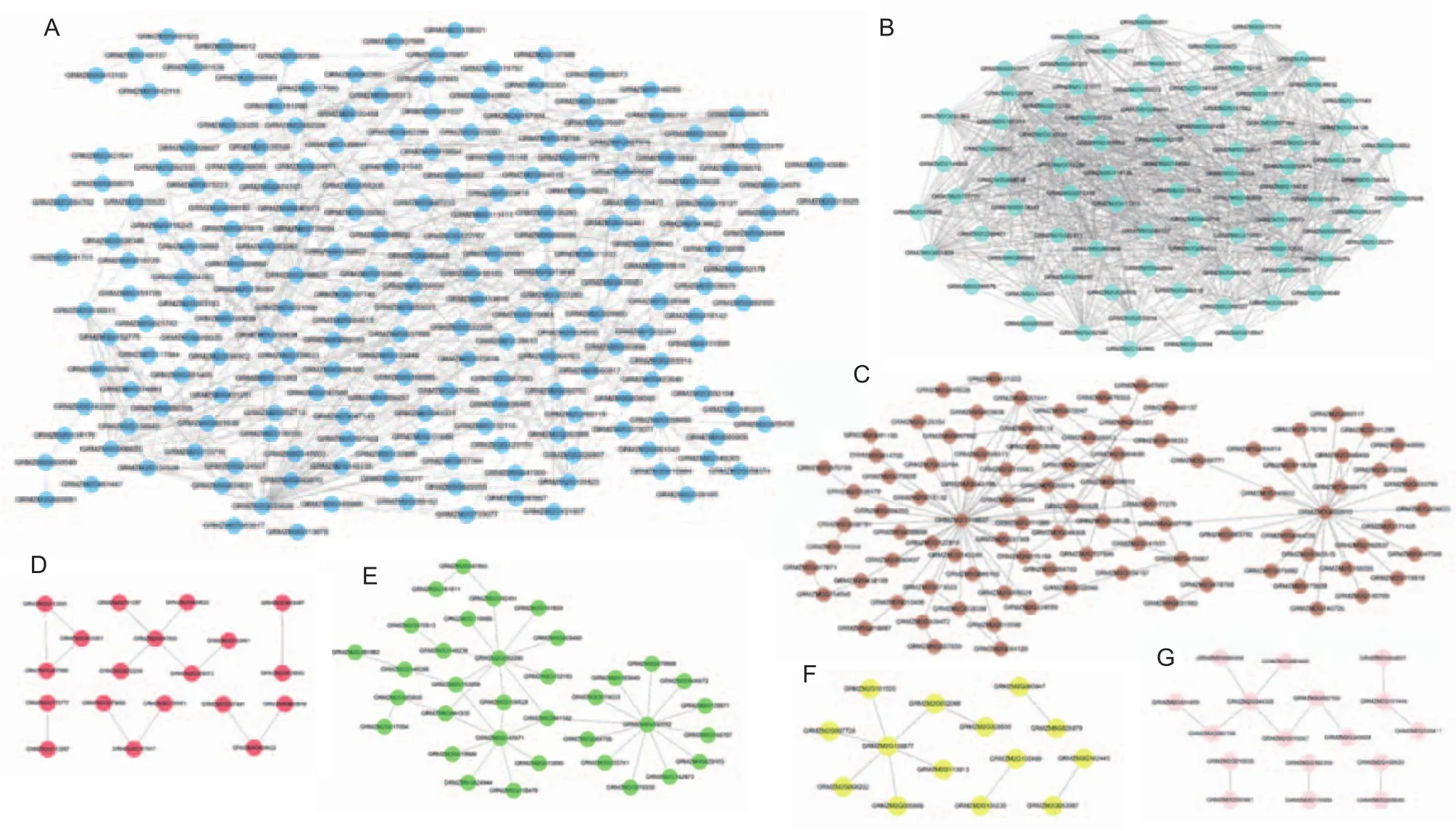
Fig. 4 The networks only composed by module genes in each module. A, Blue module network. B, Turquoise module network. C,Brown module network. D, Red module network. E, Green module network. F, Yellow module network. G, Pink module network.
For the trait TimePoint, GRMZM2G000052(LOC101027113), the hub gene of Green module is the most noteworthy gene. GRMZM2G000052 is the protein coding gene of lysine-specific histone demethylase 1. In KEGG,GRMZM2G047855 is found in the pathways “Arginine and proline metabolism, conserved biosystem” and “beta-Alanine metabolism, conserved biosystem”. GRMZM2G002100(MPK7) is the hub gene of Pink module. It is a protein coding gene involved in ABA stimulation of MAP kinase, and has been reported in multiple studies of gene functions (Zonget al. 2009). Zonget al. (2009) suggested thatZmMPK7is responsible for the removal of reactive oxygen species.Their results indicated that endogenous H2O2may be required for ZmMPK7-mediated ABA signaling, and showed that the ZmMPK7 protein located in the nucleus (Zonget al.2009). Moreover, they reported that GRMZM2G002100 is found in the KEGG pathways “MAPK signaling pathwayplant, organism-specific biosystem” and “Plant hormone signal transduction, organism-specific biosystem.”
In addition to the important genes described above,some pathways obtained by the functional enrichment analysis were also significantly associated with our two traits of interest. For instance, among the pathways of gene enrichment in the Blue module with strong correlations with LeafRegion, the most significant and strongest one was the GO term “DNA replication” (GO:0006260). DNA replication refers to the replication process of DNA double strands before cell division. It is the basis of biological inheritance, and it is also the most fundamental process of all organisms. Another significant pathway is the KEGG pathway “Spliceosome” (zma03040). Spliceosome is a multicomponent complex formed when RNA splicing occurs.Its size is 60S, and is mainly composed of small molecules of nuclear RNA and proteins. Besides, pathway “Spliceosome”(zma03040) also appeared in the Red module that was significantly associated with LeafRegion. In addition,there were several other pathways that were shared by two different modules, for instance, the KEGG pathways“ribosome” (zma03010, shared by modules Turquoise and Red) and “glutathione metabolism” (zma00480, shared by modules Turquoise and Black), and the GO term “translation”(GO:0006412, shared by modules Turquoise and Red). It shows that the correlation between these pathways and LeafRegion is more reliable (Table 2).
Of the module most correlated with TimePoint, the Green module, most of the pathways obtained in this module were related to protein and amino acid processing.Among the pathways, “Benzoxazinoid biosynthesis”(zma00402) is related to the biosynthesis of Benzoxazinoid.Benzoxazinoids (hydroxamic acids) as a kind of plant secondary metabolites could be found in the grass family and some eudicot families. And in vacuoles, glucosides are the major storage form of the primary benzoxazinoids, such as diisobutyl-n-octylamine (DIBOA) and its 2,4-dihydroxy-7-methoxy-1,4-benzoxazin-3-one (7-methoxy derivative DIMBOA). The biosynthesis of benzoxazinoid in maize occurs at a branch point from the tryptophan biosynthesis pathway where an enzyme homologue of a tryptophan synthase, BX1 (indole-3-glycerophosphate lyase), catalyzes the conversion of indole-3-glycerophosphate to indole(Kanehisa 2002). Besides, KEGG pathways “biosynthesis of antibiotics” (zma01130) and “glycolysis/gluconeogenesis”(zma00010) were shared by multiple modules that with high correlation with TimePoint, which enhanced the credibility of the correlation between these pathways and TimePoint(Table 2).
Brown module was significantly associated with LeafRegion as well as TimePoint. The representative pathways of the Brown module is mainly related to the synthesis and metabolism of biological substances, such as “citrate cycle” (TCA cycle) (zma00020), “biosynthesis of antibiotics” (zma01130), “biosynthesis of amino acids” (zma01230), and “ubiquitin mediated proteolysis”(zma04120). “Carbon metabolism” (zma01200) was the most significant pathway enriched by the module genes.Thus, we inferred that carbon metabolism of maize leaves are not only affected by the region of leaf in which the carbon is located but also by the time of day.
Through WGCNA, we obtained the variables (LeafRegion and TimePoint) related to the modules and the module eigen-genes. While methods of generating molecular coexpression networks is not new, few approaches provide such a complete network research tool as WGCNA. The implementation of WGCNA in R is user-friendly and easy to carry out. It facilitates data analysis by allowing researchers to have greater flexibility and control over the processing procedure. Moreover, coupling WGCNA with subsequent functional enrichment and gene-gene interaction analyses,aids further exploration and interpretation of the biological significance of modules and genes. However, due to the limited sample size, the findings in our study may be slightly biased. Nonetheless, as more data are made available and progress has been made on such research, we endeavor to incorporate any new and relevant findings in our future studies.
5. Conclusion
We applied the WGCNA on a set of gene expression pro files of carbon metabolism in maize leaves to identify the modules and genes that may be affected by the variables,LeafRegion and TimePoint. Our study provides an example of how this practical network analysis tool can be very useful for biologists who undertake studies based on large,multidimensional, and incompletely annotated datasets. We have elucidated potential biological relationships within each module to help improve our understanding of the module genes and pathways with the highest correlations with LeafRegion and TimePoint. The information we obtained can help direct future studies narrow down particular relationships of significance to conduct more in-depth research. The results obtained in our study can not only augment the findings of similar studies, but also provide a new framework on which researchers can develop future studies.
Acknowledgements
This study was funded by the National Nature Science Foundation of China (31671577), the Natural Science Foundation of Beijing, China (5174033), the Scientific and Technological Innovation Capacity Construction Project of Beijing Academy of Agricultural and Forestry Sciences,China (KJCX20170404), the Scientific and Technological Innovation Team of Beijing Academy of Agricultural and Forestry Sciences, China (JNKYT201604), and the Beijing Postdoctoral Research Foundation, China (2016 ZZ-66).
Appendicesassociated with this paper can be available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
 Journal of Integrative Agriculture2019年2期
Journal of Integrative Agriculture2019年2期
- Journal of Integrative Agriculture的其它文章
- Modelling and mapping soil erosion potential in China
- Updating conventional soil maps by mining soil–environment relationships from individual soil polygons
- Spatial variability of soil total nitrogen, phosphorus and potassium in Renshou County of Sichuan Basin, China
- Spatial variability of soil bulk density and its controlling factors in an agricultural intensive area of Chengdu Plain, Southwest China
- An integrated method of selecting environmental covariates for predictive soil depth mapping
- Remotely sensed estimation and mapping of soil moisture by eliminating the effect of vegetation cover