
银屑病的发病机制及其研究进展
2019-01-24 09:03程龙龙姜述堃刘技辉
医学综述 2019年2期
程龙龙,姜述堃,杜 阳,刘技辉※
(1.中国医科大学法医学院,沈阳 110122; 2.沈阳市第七人民医院皮肤科,沈阳110003)
银屑病,俗称牛皮癣,其组织病理学表现主要为表皮基底层角质形成细胞角化过度伴角化不全,真皮层毛细血管增生、扩张,炎性细胞浸润,最常见的临床表现为红色或粉色的斑块、鳞屑等[1]。关于银屑病的发病机制目前尚无定论,细胞免疫系统异常与角质形成细胞异常是银屑病皮损组织的两大特征性表现,也是银屑病发病机制研究的主要切入点[2]。其中,辅助性T细胞(helper T cell,Th)17淋巴细胞及其细胞因子、角质形成细胞的信号转导通路与角质形成细胞中微RNAs(microRNAs,miRNAs)表达水平等是近年来的研究热点。此外,血管内皮生长因子(vascular endothelial growth factor,VEGF)也在银屑病发病过程中起重要作用。以相关细胞因子、miRNAs为治疗靶点,对免疫系统和角质形成细胞内的基因表达进行调控是银屑病治疗的重要策略。根据不同靶点的治疗效果,可相互印证各靶点在银屑病发病过程中的作用与影响,从而进一步阐明其发病机制。现就银屑病的发病机制及其研究进展予以综述。
1 免疫细胞功能紊乱及其相关细胞因子水平异常

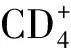
有学者发现,IL-9也参与Th17细胞、Treg细胞的分化与调节[16]。Singh等[17]研究发现,银屑病患者外周血中Th9细胞百分比及IL-9水平明显高于正常人,且与银屑病严重程度(PASI评分)显著相关。Chowdhury等[18]认为,抑制IL-9可能对银屑病发病中的致病因子产生广泛影响,从而可对银屑病的进展进行控制。
目前,大量临床及细胞实验证实,Th17细胞与Treg细胞及其细胞因子的免疫功能异常是导致自身免疫性疾病的发病机制之一[19-21]。而与Th17细胞与Treg细胞相关的免疫细胞因子可作为相关治疗靶点,为控制银屑病的发生发展提供新思路。
2 皮损区VEGF水平异常升高
一般认为,银屑病的特征性水肿、红斑等表现是由于真皮血管扩张、通透性升高等异常反应引起。VEGF属于酪氨酸激酶家族成员,其与表达在血管内皮细胞上的两个Ⅲ型酪氨酸激酶受体(VEGF受体1与VEGF受体2)结合作用于内皮细胞,从而促进真皮血管内皮细胞增殖并增加血管的通透性。在角质细胞中,VEGF与受体结合后激活p21活化激酶1信号通路,而p21活化激酶1活化后可诱导细胞异常增殖、细胞迁移活性增强,并通过调节VEGF的表达促进银屑病患者皮损组织中角质形成细胞生物学行为改变及新生血管形成,从而参与银屑病的发病过程[22]。Jiang等[23]通过在小鼠皮肤局部注射VEGF证实,其与银屑病的角蛋白异常表达密切相关。在皮肤组织中,角质形成细胞分泌VEGF,后者通过旁分泌作用促进皮损组织局部血管内皮细胞增殖、迁移,也可通过自分泌作用影响角质形成细胞。
既往研究发现,VEGF过表达并不能立即表现出银屑病皮损现象,随着年龄增加银屑病症状(Koebner征)才会出现并加重[24]。Sudhesan等[25]对印度南部银屑病患者的VEGF基因进行多态性分析发现,VEGF基因是患银屑病的风险因素,且银屑病患者血清中的VEGF水平明显高于正常人,表明VEGF在皮肤组织一定时期的过多表达会导致银屑病的发生。Chen等[26]通过5-氨基酮戊酸光动力疗法(5-aminolevulinic acid-photodynamic therapy, ALA-PDT)对角蛋白14-VEGF转基因小鼠进行治疗发现,其可以削弱小鼠皮损组织中的炎性细胞浸润和降低微血管密度,改善临床表现,在一定程度上阻止银屑病的发展。虽然VEGF是维持银屑病活动性的关键因素,但与疾病的严重程度(PASI评分)并无直接关系。Liu等[27]的研究结果显示,萨力多胺可以降低咪喹莫特诱发的银屑病模型小鼠皮肤中的VEGF和肿瘤坏死因子(tumor necrosis factor,TNF)α水平,改善银屑病皮损组织的病变,但其仅仅降低皮损组织中的VEGF水平,对正常皮肤组织的VEGF水平影响不明显。
3 角质形成细胞中的信号转导通路异常
角质形成细胞中的细胞因子可通过多种信号转导通路进行细胞内信号转导,包括Ca2+依赖的蛋白激酶C途径、促分裂原活化的蛋白激酶途径等,其中Janus激酶-信号转导及转录激活因子(Janus kinase-signal transduction and activator of transcription, JAK-STAT)信号转导途径是目前银屑病的研究热点。
JAK-STAT通路是多数细胞因子最重要的胞内信号转导通路,细胞因子与靶细胞上相应的受体相结合并激活JAK,JAK磷酸化相应的STAT,活化的STAT将信号由细胞膜传至细胞核,从而发挥多种生物学效应。Orecchia等[28]研究证实,磷酸化的STAT3是诱导小鼠表皮组织实验性癌变的必要条件,且适度连续的STAT3磷酸化所形成的表皮组织增生与促炎因子刺激所形成的表皮组织增生相似。Kim等[29]用苦参黄素治疗银屑病模型小鼠发现,其可以直接抑制JAK-STAT通路,从而抑制促炎介质的表达。Galluzzo等[30]研究表明,托法替尼可以竞争性抑制ATP与JAK结合,阻止下游的STAT活化,使STAT无法调节银屑病相关的炎性介质基因,从而阻断银屑病进程。Nada等[31]用NB-UVB照射疗法对银屑病、白斑患者进行系统治疗后发现,治疗前后银屑病、白斑患者体内的JAK1水平明显下降,表明抑制JAK与STAT的高表达可有效缓解银屑病的症状。
此外,细胞因子信号转导抑制物(suppressor of cytokine signaling,SOCS)作为JAK/STAT信号转导通路的主要负调节因子,通过负反馈方式调节JAK/STAT信号转导通路的抑制物[32]。其发挥作用的常见机制为通过抑制SOCS1促进相关趋化因子高表达,使表皮组织中的炎性细胞趋化聚集,进而加重银屑病皮损的严重程度[33]。Madonna等[34]研究发现,在维持抗凋亡的过程中,SOCS1与SOCS3在银屑病角质形成细胞中大量表达,其与银屑病皮损区强烈的不良反应密切相关。Ahmed等[35]发现,在SOCS1和SOCS3的结构上有一种叫作激酶抑制区域的12种氨基酸残基的关键功能域,其在抑制JAK2酪氨酸激酶的过程中发挥一定作用。在银屑病患者皮损区的角质形成细胞中,高水平的SOCS可通过抑制JAK-STAT通路下调促炎因子水平,如TNF-β、γ干扰素等,进而降低免疫系统的损害效应。但相关研究表明,仅靠SOCS并不能完全阻断JAK-STAT通路在银屑病发病过程中的作用[34-37]。
多种瞬时感受器电位(transient receptor potential,TRP)为无选择性离子通道,在皮肤疾病的发病过程中发挥重要作用。研究表明,多种TRP通道参与调控机体对疼痛、瘙痒和发热的反应过程,参与皮肤屏障的形成和维护、皮肤细胞和器官生长分化、皮肤的免疫和炎症过程等[38-39]。TRP通道在控制细胞胞内稳态、调节细胞结局、免疫和炎症机制及相关细胞因子的分泌过程等方面发挥不可或缺的作用[40-41]。银屑病皮损组织基底层钙离子水平较高,且含有较少的细胞外钙,说明银屑病皮损组织丧失了正常的钙离子梯度[42]。在角质形成细胞的分化与增殖过程中,异常的钙离子梯度可能是银屑病发病的机制之一。在体外试验中,银屑病患者的角质形成细胞在TRPC6活化剂的催化下部分完善了细胞的分化及增殖缺陷,而角质形成细胞外髓鞘的TRPV1促使部分细胞因子表达增加,说明TRP通道与炎症反应之间存在双向干扰[43]。Leuner等[44]发现,在银屑病患者皮肤组织活检中,角质形成细胞中TRPC通道的信使RNA及相关蛋白表达减少,分化标志物水平降低,提示分化机制受损,此类缺陷在银屑病患者的皮损组织及非皮损组织中均存在。此外,TRP通道(TRPC5、TRPC5、TRPM4、TRPM7通道)通过调节T细胞释放的细胞因子,参与相关炎症反应的发生。Huang等[45]研究发现,黄芩素通过激活TRPV4受体诱导钙离子,促进角蛋白1、角蛋白10表达,表明黄芩素具有促进角质细胞分化、抑制增殖的作用。目前,TRP通道作为一种调节组织内钙离子水平的重要途径,有望成为一个缓解银屑病症状、治疗银屑病的新靶点。
4 PSORSs基因、基因组甲基化与miRNAs的异常表达
流行病学和遗传学研究发现,银屑病是一种多基因控制的遗传性疾病[46-48]。国内外研究团队应用连锁分析的方法,在全基因组范围内发现10个银屑病易感基因位点,分别命名为PSORS1(6p)、PSORS2(17q)、PSORS3(4q)、PSORS4(1q)、PSORS5(3q)、PSORS6(19p)、PSORS7(1p)、PSORS8(16q)和 PSORS9(4q) 及PSORS10(18p)[48-51]。其中,IL-12B基因、IL-23信号通路的3个基因(IL-23A、IL-23R和IL-12B)、TNF-α下游调节核因子κB表达的两个基因(TNIP1和TNFAIP3)、调节Th2型免疫反应的两个基因(IL-4和IL-13)及晚期角质化包膜(late cornified envelope,LCE)基因簇中的LCE3A和LCE3D等均与银屑病显著相关,提示免疫系统和基因变异在银屑病发病过程中起重要作用[52]。Austin等[53]报道,花青素可有效上调LCE在角质形成细胞中的表达,在治疗银屑病方面具有潜在作用。Niehues等[54]通过对三维皮肤模型进行定量特征轨迹分析发现,LCE3A蛋白在特定水平下具有抗菌活性,且在表皮宿主防御和LCE3型银屑病的发病过程中发挥特殊作用。
此外,与正常人相比,寻常型银屑病患者的基因组甲基化水平升高,且免疫组织化学结果显示,寻常型银屑病患者皮损组织中的抗5-甲基胞嘧啶表达水平也升高[55]。Zhang等[56]对银屑病患者皮损区和非皮损区进行甲基化DNA免疫测序分析发现,寻常型银屑病患者皮损组织/非皮损组织中基因PDCD5和金属蛋白酶组织抑制因子2(tissue inhibitors of metalloproteinases-2,TIMP-2)的信使RNA表达水平异常,且与DNA甲基化水平呈负相关,说明基因PDCD5和TIMP2的甲基化水平(基因的表达水平)与寻常型银屑病患者皮损状态(PASI评分)有关,故DNA甲基化水平可以作为判断银屑病严重程度的指标之一。Gaewska等[57]研究发现,与正常人皮肤相比,银屑病皮损区的基质金属蛋白酶3、基质金属蛋白酶9、TIMP-2的血浆水平均显著上升;与非皮损区相比,TIMP-2信使RNA在皮损区表达水平升高,且呈低甲基化状态,提示基因TIMP-2在寻常型银屑病非皮损区可能发挥抑制角质形成细胞过多增殖,阻止其向银屑病皮损方向发展的作用。
差异性miRNAs所调控的基因可能通过影响促分裂原活化的蛋白激酶信号通路在细胞增殖、炎症形成、血管增生、体液免疫等方面参与银屑病的发病过程[58]。Chowdhari等[59]研究证实,银屑病患者皮损区的miR-4516表达水平降低会加速细胞迁移,进而抵抗细胞凋亡和分化,同时也强调了它可能用于银屑病治疗干预的潜在价值。人类角质形成细胞中miR-4516下调可导致细胞骨架(Rac1、RhoA、Cdc42)重组,从而显著抑制细胞的增殖与分化。Hermann等[60]研究也发现,银屑病皮损区miR-146a、miR-146b的表达均增加,且miR-146a在角化形成细胞中对促炎因子更敏感。目前,miRNAs广泛存在于多个物种中,在基因组学上的应用为银屑病的基因调控提供了许多有效治疗的优势,但由于数量众多,各miRNAs之间调节通路复杂多样,故仍有很多问题亟待解决。
5 小 结
银屑病是一种由环境因素刺激、多基因遗传控制、免疫机制介导的皮肤病,其发病机制复杂,在其发生发展过程中涉及血管增生迁移、天然T细胞分化、炎性细胞浸润、角质形成细胞增殖等多个环节。目前认为,免疫细胞活化与角质形成细胞分化异常是银屑病发病的关键,遗传、感染、代谢障碍、内分泌异常及其他相关因素,包括外伤、精神、维生素D缺乏和吸烟等均可诱发银屑病易感者发病和加重银屑病。目前为止,尚未有任何一种药物能够完全治愈银屑病或控制复发。未来,需进一步分析各因素(Th17细胞、STAT3、PSORSs基因等)在银屑病发病过程的具体作用及相互关系,以为银屑病的靶向治疗提供依据。
猜你喜欢
云南医药(2021年3期)2021-07-21
儿童故事画报·智力大王(2020年1期)2020-04-28
文物保护与考古科学(2016年1期)2016-04-16
医学研究杂志(2015年4期)2015-06-10
中国医学科学院学报(2015年5期)2015-03-01
中国当代医药(2015年32期)2015-03-01
中国医药导报(2015年24期)2015-02-28
中国医疗美容(2015年5期)2015-02-03
中华皮肤科杂志(2014年4期)2014-12-19
中华皮肤科杂志(2014年3期)2014-12-19
