
咳喘六味合剂质量标准提高研究
2019-01-24 09:39杨帆平李国文
药学实践杂志 2019年1期
杨帆平,李国文,奚 燕
(1.上海中医药大学附属普陀医院药物临床试验机构办公室,上海 200062;2.上海中医药大学附属上海市中西医结合医院药剂科,上海 200082;3.上海中医药大学附属龙华医院药剂科,上海 200032)
咳喘六味合剂是以中医药理论为指导,结合临床实践,将麻黄、附子、黄芩、细辛、桃仁和虎耳草配制而成的院内制剂,该药温阳抗寒、化痰平喘,主要用于虚寒型或是寒邪较盛的咳喘治疗。哮喘属于中医“哮病”范畴,长期临床经验表明,中医药在治疗哮喘方面具有优势,本方较好地体现了祖国医学“治病求本”的思想[1]。
咳喘六味合剂适用于肾阳虚哮喘患者,临床疗效显著[2],但是其质量标准的制订时间较早,且仅针对麻黄一种药味进行了定性鉴别,对外观、pH值等检查项缺少有效的含量测定方法。本实验增加了咳喘六味合剂中的黄芩、细辛的薄层色谱(TLC)鉴别方法,新建了麻黄、黄芩的含量测定方法,并进行了方法学验证,为提高和完善咳喘六味合剂的质量标准提供了可靠依据。
1 仪器和试药
Sartorius BS110S分析天平(万分之一级);Sartorius CP225D分析天平(十万分之一级);硅胶G薄层预制板(青岛海洋化工厂);其余试剂均为分析纯;对照品:黄芩苷(批号:110715-201318),黄芩素(批号:111595-201607),汉黄芩素(批号:111514-201605),细辛脂素(批号:111889-201504),盐酸麻黄碱(批号:171241-201508),盐酸伪麻黄碱(批号:171237-201509),黄芩中药材(批号:120955-201309),细辛中药材(批号:121204-201405),以上对照品均由中国食品药品检定研究院提供。咳喘六味合剂供试品(批号:160918),龙华医院制剂室。
2 方法与结果
2.1 黄芩的薄层鉴别
2.1.1供试品溶液的制备
精密量取咳喘六味合剂5 ml置具塞锥形瓶中,加乙酸乙酯-甲醇(3∶1)30 ml,超声处理30 min,滤过,蒸干,残渣加甲醇5 ml使溶解,摇匀,作为供试品溶液。
2.1.2对照药材溶液的制备
取黄芩对照药材0.5 g,加乙酸乙酯-甲醇(3∶1)30 ml,按“2.1.1”项下方法制成对照药材溶液。
2.1.3对照品溶液的制备
取黄芩苷、黄芩素、汉黄芩素对照品,加甲醇溶解,制成每1 ml含1 mg的对照品溶液。
2.1.4阴性样品溶液的制备
取除黄芩药材外的其他五味药材按相同处方配比和工艺制备阴性样品,取5 ml,再按“2.1.1”项下方法制得阴性样品溶液。
2.1.5薄层条件和结果
按照TLC法[3](《中华人民共和国药典》2015年版四部通则0502)实验,吸取供试品溶液、对照药材溶液、阴性样品溶液各2 μl以及对照品溶液1 μl,分别点于同一聚酰胺薄膜板上,以甲苯-乙酸丁酯-甲醇-甲酸 (10∶3∶1∶2)为展开剂,预饱和30 min,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果如图1所示,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点;在与对照品色谱相应的位置上,显3个相同颜色的斑点,且阴性样品溶液对结果没有影响。说明本方法可用于鉴别咳喘六味合剂中的黄芩。
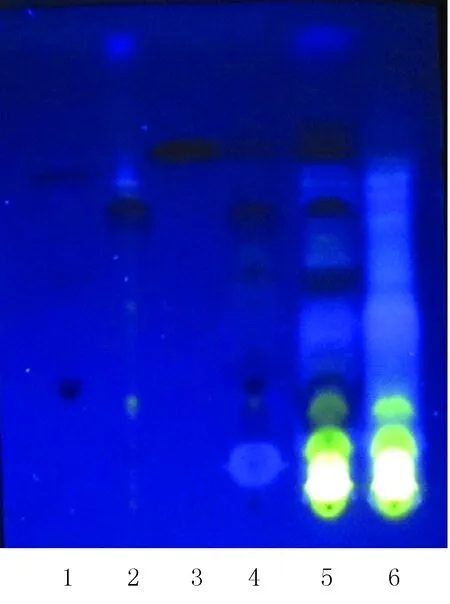
图1 黄芩薄层鉴别图1.黄芩苷;2.黄芩素;3.汉黄芩素;4.黄芩对照药材;5.供试品;6.阴性样品
2.2 细辛的薄层鉴别[4]
2.2.1供试品溶液的制备
取咳喘六味合剂10 ml,加甲醇50 ml,加热回流30 min,滤过,蒸干,残渣加水30 ml使溶解,加石油醚(60~90 ℃)振摇提取2次,每次20 ml,合并石油醚液,挥干,残渣加乙酸乙酯0.5 ml,使溶解,摇匀,作为供试品溶液。
2.2.2对照药材溶液的制备
取细辛对照药材0.5 g,加甲醇50 ml,按“2.2.1”项下方法制得对照药材溶液。
2.2.3对照品溶液的制备
称取细辛脂素对照品,加甲醇溶解制成每1 ml含1 mg的对照品溶液。
2.2.4阴性样品溶液的制备
取除细辛药材外的其他五味药材,按相同处方配比和工艺制备阴性样品,取10 ml,按“2.2.1”项下方法制得阴性样品溶液。
2.2.5薄层条件和结果
按照TLC法[3]实验,吸取上述溶液各15 μl,分别点于同一硅胶G板上,以环己烷-三氯甲烷-乙酸乙酯(16∶3∶4)为展开剂,展开,取出,晾干,喷5%香草醛硫酸溶液,105 ℃加热至斑点显色清晰。结果如图2所示,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的斑点,阴性样品溶液对结果无影响。说明本方法可用于鉴别咳喘六味合剂中的细辛。
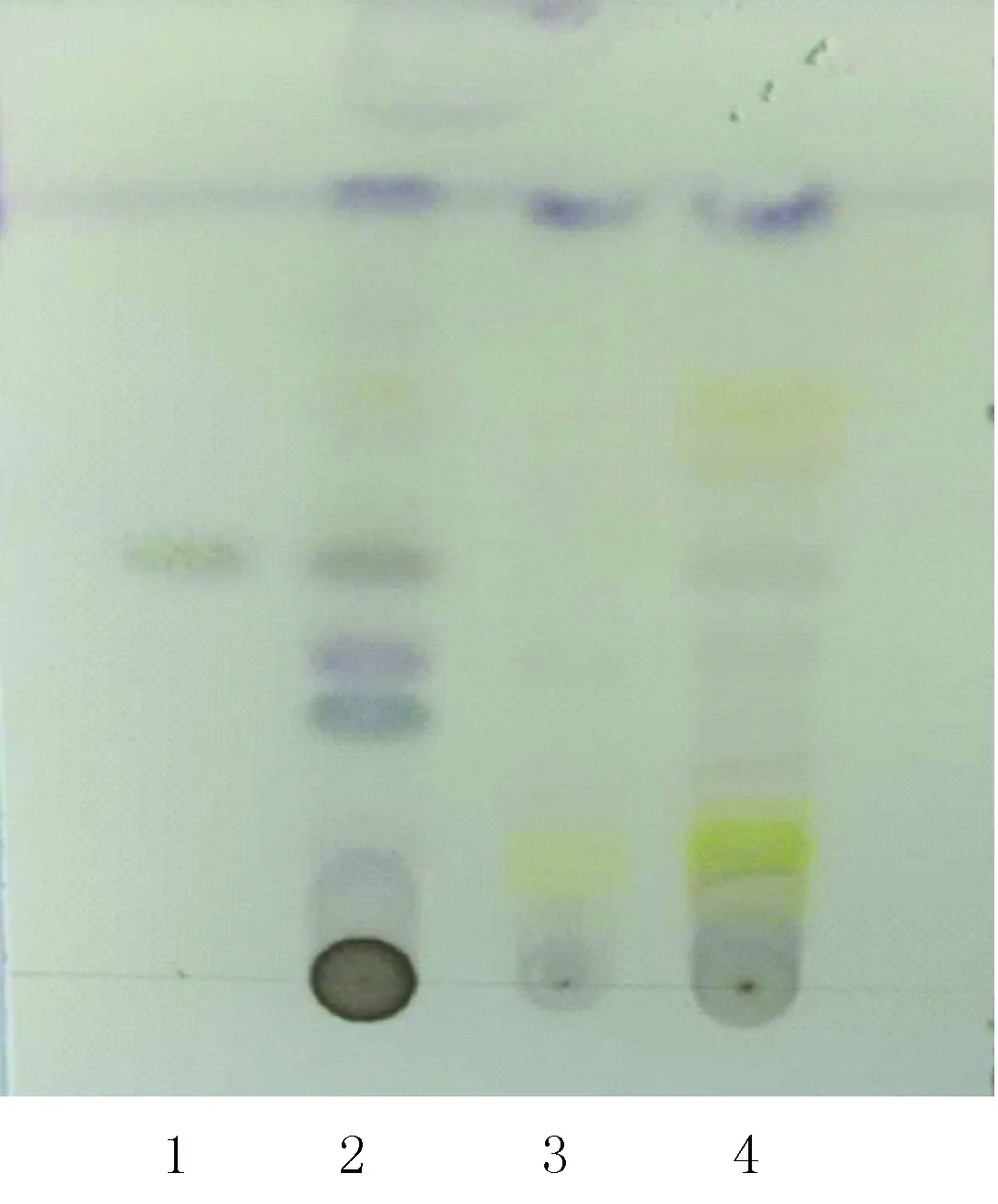
图2 细辛薄层鉴别图1.细辛脂素;2.对照药材;3.阴性样品;4.供试品
2.3 麻黄的含量测定[5]
2.3.1色谱条件与系统适用性试验
以十八烷基硅烷键合硅胶为填充剂;乙腈-0.1%磷酸溶液(4∶96)为流动相;检测波长206 nm;流速1.0 ml/min。理论板数按盐酸麻黄碱峰计算,应不低于7 000。
2.3.2对照品溶液的制备
取盐酸麻黄碱对照品15 mg,精密称定置于50 ml量瓶中,甲醇使之溶解,定容至刻度,摇匀;另精密称定盐酸伪麻黄碱对照品25 mg,以同法制成溶液;分别精密量取上述溶液5、2 ml,置同一25 ml量瓶中,流动相:乙腈-0.1%磷酸溶液(4∶96),稀释至刻度,摇匀,即得。
2.3.3供试品溶液的制备
精密量取咳喘六味合剂2 ml,置分液漏斗中,加水10 ml及浓氨试液1 ml,摇匀,乙醚液振摇提取3次,每次30 ml,合并乙醚液,加5%盐酸甲醇溶液5 ml,摇匀,40 ℃回收溶剂至干,残渣加上述流动相溶解,并转移至25 ml量瓶中,稀释至刻度,摇匀,即得。
2.3.4阴性样品溶液的制备
取按处方去除麻黄制得的阴性样品2 ml,按“2.3.3”项下方法制得阴性样品溶液。
2.3.5专属性试验
精密吸取上述对照品、供试品、阴性样品溶液各10 μl,分别注入液相色谱仪,进行测定。结果表明,在本实验选用的色谱条件下,供试品与对照品溶液在相同保留时间出峰,阴性样品溶液对盐酸麻黄碱和盐酸伪麻黄碱的测定无干扰(图3)。

图3 盐酸麻黄碱、盐酸伪麻黄碱HPLC图A.对照品;B.阴性样品;C.供试品;1.盐酸麻黄碱;2.盐酸伪麻黄碱
2.3.6线性关系考察
分别精密称取盐酸麻黄碱、盐酸伪麻黄碱对照品15.05、9.97 mg,置同一50 ml量瓶中,加上述流动相溶解,稀释至刻度,摇匀。分别精密量取上述混合溶液2、5、10、25 ml,分别置50 ml量瓶中,加上述流动相稀释至刻度,摇匀。精密吸取制得的溶液及母液各10 μl,注入液相色谱仪,测定。以对照品浓度为横坐标,以峰面积积分值为纵坐标,绘制标准曲线,计算回归方程。结果显示,盐酸麻黄碱在12.04~301.00 μg/ml范围内呈现良好的线性关系,回归方程为Y=23.519 8X-10.400 8,相关系数r=1.000 0;盐酸伪麻黄碱在7.98~199.40 μg/ml范围内呈现出良好的线性关系,回归方程为Y=23.986 4X-8.284 7,r=1.000 0。
2.3.7稳定性试验
制备供试品溶液,分别在6个时间点(0、2、4、8、12、24 h)测定峰面积,每次精密吸取10 μl进样。结果得到盐酸麻黄碱在6个时间点的峰面积积分值RSD为0.2%,盐酸伪麻黄碱的RSD为1.3%,说明咳喘六味合剂在24 h内稳定。
2.3.8重复性试验
制备6份供试品溶液,分别进样10 μl,测定含量,结果盐酸麻黄碱、盐酸伪麻黄碱的平均含量分别为0.84 mg/ml和0.62 mg/ml,RSD分别为0.11%和0.92%,结果表明本方法重复性较好。
分别由两人各自配制相同浓度的供试品溶液6份,通过不同的仪器和试剂,计算12个含量数据的相对标准差。结果得到盐酸麻黄碱含量的RSD为0.21%,盐酸伪麻黄碱含量的RSD为1.1%,表明试验符合要求。
2.3.9回收率试验
分别精密称取盐酸麻黄碱、盐酸伪麻黄碱对照品18.32、15.07 mg,置同一50 ml量瓶中,用上述流动相溶解,稀释至刻度,混匀,备用;精密量取咳喘六味合剂1 ml,共9份,置分液漏斗中,分别加入上述配制好的对照品溶液0.8、1.0、1.2 ml,平行3份,分别加水10 ml及浓氨试液1 ml,摇匀,乙醚液振摇提取3次,每次30 ml,合并乙醚液,加5%盐酸甲醇溶液5 ml,混匀,40 ℃回收溶剂至干,残渣加流动相溶解,转移至25 ml量瓶中,稀释至刻度,摇匀,即得;计算回收率得到,盐酸麻黄碱平均回收率为101.7%,RSD为1.5%;盐酸伪麻黄碱平均回收率为101.6%,RSD为2.4%,结果表明本方法准确度高,回收率较好。
2.4 黄芩的含量测定
2.4.1色谱条件与系统适用性试验
以十八烷基硅烷键合硅胶为填充剂;甲醇-0.1%磷酸溶液(47∶53)为流动相;检测波长278 nm;流速1.0 ml/min。理论板数按黄芩苷峰计算,应不低于5 000。
2.4.2对照品溶液的制备
取黄芩苷对照品15 mg,精密称定,置50 ml量瓶中,加甲醇溶解,稀释至刻度,摇匀;精密量取5 ml,置50 ml量瓶中,加甲醇稀释至刻度,摇匀,即得。
2.4.3供试品溶液的制备
精密量取咳喘六味合剂5 ml,置50 ml量瓶中,加50%甲醇稀释至刻度,摇匀;精密量取2 ml,置50 ml量瓶中,加50%甲醇稀释至刻度,摇匀,滤过,即得。
2.4.4阴性样品溶液的制备
取按处方去除黄芩制得的阴性样品5 ml,按“2.4.3”项下方法制得阴性样品溶液。
2.4.5专属性试验
精密吸取上述对照品、供试品、阴性样品溶液各10 μl,分别注入液相色谱仪,进行测定。结果表明,在本实验选用的色谱条件下,供试品与对照品溶液在相同保留时间出峰,阴性样品溶液对黄芩苷的测定无影响(图4)。

图4 黄芩苷HPLC图A.对照品;B.阴性样品;C.供试品;1.黄芩苷
2.4.6线性关系考察
精密称取黄芩苷对照品13.88 mg,置50 ml量瓶中,甲醇溶解,稀释至刻度,摇匀。分别精密量取1、2、3、5、6、10 ml,分别置50 ml量瓶中,用流动相甲醇-0.1%磷酸溶液(47∶53)稀释至刻度,摇匀。以上溶液及母液各精密吸取10 μl,分别注入液相色谱仪,测定。以黄芩苷对照品浓度为横坐标,以峰面积积分值为纵坐标,绘制标准曲线,计算回归方程。结果显示黄芩苷在5.18~129.50 μg/ml范围内呈现良好的线性关系,回归方程为Y=39.015 1X-15.643 9,r=0.999 9。
2.4.7稳定性试验
制备供试品溶液,分别在7个时间点(0、2、4、8、12、18、24 h)测定峰面积,每次精密吸取10 μl进样。结果得到7个时间点测定的峰面积积分值RSD为0.4%,表明咳喘六味合剂在24 h内稳定性良好。
2.4.8重复性试验
制备6份供试品溶液,分别进样10 μl。通过计算得到,黄芩苷的平均含量为9.0 mg/ml,RSD为1.0%,结果表明本方法的重复性良好。
分别由两人各自配制相同浓度的供试品溶液6份,通过不同的仪器和试剂,计算12个含量数据的相对标准差。结果得到样品含量的RSD为1.3%,表明试验符合要求。
2.4.9回收率试验
精密量取咳喘六味合剂2.5 ml,共9份,分别置于50 ml量瓶中,以3份为一组,每组分别加入黄芩苷对照品约18、 22.5、27 mg,甲醇溶解,稀释至刻度,摇匀;精密量取5 ml,置50 ml量瓶中,加甲醇稀释至刻度,摇匀。计算平均回收率为101.0%,RSD为0.3%,结果表明本方法准确度较高。
3 讨论
咳喘六味合剂中的附子是一味常用的温里药,生药材使用前需经炮制减毒,其含有的主要成分可为单酯型和双酯型生物碱。笔者尝试用HPLC法对其进行定量测定,但阴性样品干扰较大,不能准确控制其含量,后续将对此开展研究。虎耳草属于民族医药,未被《中国药典》收载,只收录于贵州省地方药材标准。现有研究已发现其乙醇提取物中含有多元酚、黄酮、有机酸等[6]多种成分,但由于研究时间较短,需要进一步探索其活性部位的活性成分。
展开剂的选择对TLC中待鉴别成分的分离效果至关重要。对咳喘六味合剂中的桃仁,笔者曾尝试以三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)5~10 ℃放置12 h的下层溶液为展开剂进行TLC研究,但无论是日光还是荧光下,都未能在与对照药材色谱相应位置上显示相同颜色斑点。而在对细辛的鉴别过程中,笔者采取了两种不同的方法,其中,以石油醚(60~90 ℃)-乙酸乙酯(3∶1)为展开剂的方法未能成功分离得到满意的结果。
对于麻黄中主要药效成分的含量测定,《中国药典》(2015年版)标明的测定方法中,色谱柱为乙醚键合硅胶C18柱,而本实验选用了常规的C18柱对其进行含量测定,结果发现两者的分离度均大于2.0,理论塔板数超过10 000,比药典方法更加简便易行。
在黄芩苷含量测定的实验中,对于流动相的选择,首先参照《中国药典》(2015年版)中【黄芩】项下的含量测定方法,然后根据系统适用性要求调整流动相比例,当甲醇与0.1%磷酸的比例为47∶53时,色谱峰分离度较好,理论塔板数高。
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
河北果树(2020年2期)2020-05-25
基层中医药(2018年4期)2018-08-29
中成药(2018年5期)2018-06-06
中成药(2017年7期)2017-11-22
中国民族民间医药·下半月(2014年2期)2014-09-26
中国中医药现代远程教育(2014年19期)2014-03-01
中国中医药现代远程教育(2014年17期)2014-03-01
中国中医药现代远程教育(2014年15期)2014-03-01
天津药学(2013年2期)2013-12-23
