
分子印迹固相萃取-高效液相色谱法测定植物油中11种多环芳烃
2019-01-16 01:58蕊叶金吴宇轩志宏刚王松雪马海华
中国粮油学报 2018年11期
张 冰 张 蕊叶 金吴 宇轩志宏 谢 刚王松雪马海华
(国家粮食局科学研究院1,北京 100037)(河南工业大学信息科学与工程学院2,郑州 450000)
多环芳烃(Polycyclic aromatic hydrocarbons, PAHs)是指含有两个或两个以上稠合苯环的化合物,是一类化学致癌物和环境污染物,因此国内外植物油相关标准中均对PAHs做出严格的限量规定。欧盟关于食品中多环芳烃的条例(EU) NO 835/2011[1]中规定了可直接消费或作为食品成分的油脂中苯并(a)芘的最高残留限量为2 μg/kg,同时要求苯并(a)芘、苯并(a)蒽、苯并(b)荧蒽、四种(PAH4)总量不超过10 μg/kg。GB 2716—2005 《食用植物油卫生标准》[2]也明确规定了植物油中苯并(a)芘含量应小于等于10.0 μg/kg。同时,GB 2762—2017《食品安全国家标准 食品中污染物限量》[3]对油脂及其制品中苯并(a)芘做出相同的限量规定。
植物油中主要成分甘油三酯及其脂肪伴随物严重影响着多环芳烃的分析检测,因此植物油中多环芳烃的提取净化是检测植物油中多环芳烃的关键。目前主要的提取净化方法有液液萃取法[4]、固相萃取法[5](SPE)、固相微萃取法[6](SPME)、凝胶渗透色谱法[7](GPC)等。近几年来,分子印迹聚合物(Molecularly imprinted polymers, MIPs)受到越来越多的关注。MIPs是以目标分子为模版,将具有结构互补的功能单体通过共价键或非共价方式和模版分子结合,进行聚合反应,再洗脱模版分子得到。MIPs作为固体吸附剂在固相萃取、基质固相分散萃取、固相萃取、搅拌棒吸附萃取和磁萃取等前处理中有着广泛的应用[8]。分子印迹固相萃取(molecularly imprinted polymer solid-phase extraction, MIP-SPE)通过将分子印迹技术和固相萃取技术相结合,以分子印迹聚合物为固相萃取的吸附剂,克服了传统固相萃取选择性差的缺点,对目标组分特异性的吸附结合,在生物、食品、环境等复杂样品中有很好的应用[9]。胡园等建立分子印迹固相萃取结合超高校液相荧光法测定螺中15种多环芳烃的方法,在5、10、20 μg/kg加标水平下,15种PAHs回收率73.07%~108.06%,相对标准偏差0.6%~9.2%[10]。朱琳等[11]通过高分子印迹固相萃取-气相串联质谱法测定动植物油脂中15+1种欧盟优控多环芳烃,16种PAHs回收率37.90%~105.24%,相对标准偏差小于15%。
常用的PAHs检测方法有气相串联质谱法[5]、液相串联质谱法[12]、高效液相色谱法[7]、荧光光谱法[13]等方法,其中气相串联质谱法难以应对高沸点PAHs检测,对热稳定性较差的多环芳烃检测较差。液相质谱联用仪器昂贵,对人员要求较高。荧光光谱法不适用于多组分PAHs的同时检测。而高效液相色谱-荧光检测器法,仪器较为普及,相对其他测定方法有灵敏度高,检出限低,检测种类多,分析时间短等优势,可用于包括高沸点PAHs在内的多种PAHs的同时定量检测。
1 材料与方法
1.1 材料与试剂
12种食用植物油样品,葵花籽油、菜籽油、芝麻油、大豆油、玉米油、花生油和橄榄油等,购自当地超市。
二氯甲烷、正己烷、环己烷、乙腈,色谱纯; EPA16多环芳烃混标,溶解于乙腈溶剂;SPE固相萃取柱Cleanert PAH MIP;液相色谱柱Athena PAHs HPLC Column, 4.6 × 250 mm, 5 μm。
1.2 仪器
Waters e2695高效液相色谱仪,配备荧光检测器;12位固相萃取装置。
1.3 样品处理
称取0.5 g植物油样品至15 mL离心管中,3 mL环己烷溶解,所使用容器事先均经过正己烷清洗排除本底值干扰。分子印迹柱经过10 mL二氯甲烷和5 mL环己烷活化平衡,将3 mL溶解样品的环己烷加入到固相萃取柱中,用4 mL环己烷淋洗抽干小柱,再用10 mL二氯甲烷洗脱,收集洗脱后的样品溶液氮吹至近干,1 mL乙腈定容,上机测定。
1.4 仪器条件
梯度洗脱:流动相A:乙腈,流动相B:超纯水;流速:1.2 mL/min;柱温:30 ℃;进样量:20 μL。流动相梯度条件见表1,荧光检测器荧光条件设置波长见表2。

表1 多环芳烃梯度洗脱条件

表2 多环芳烃荧光条件参数设置
2 结果与讨论
2.1 固相萃取前处理条件优化
2.1.1 淋洗体积用量的优化
本实验选用环己烷[14]作为淋洗溶剂,淋洗的主要目的在于去除甘油三酯及其他油脂伴随物,避免这些物质影响多环芳烃的分离和检测。考察了2、4、5、6、8 mL淋洗体积环己烷的淋洗效果。图1为五种不同体积的环己烷淋洗后洗脱液中干物质质量关系图。结果显示,经过不同体积环己烷淋洗后洗脱液干物质的质量分别为0.038、0.021、0.015、0.012、0.010 g,说明样品中甘油三酯及其他油脂伴随物随着淋洗体积的增大而逐渐被洗出。图2为11种多环芳烃不同淋洗体积下的回收率,2 mL淋洗多环芳烃回收率为91.89%~108.95%,重质的多环芳烃回收率低于其他淋洗体积,原因可能是重质多环芳烃与分子印迹聚合物结合力相对弱,淋洗时少量重质多环芳烃与甘油三酯一同被洗出。通过优化选择4 mL作为最优的淋洗体积用量,在保证回收率的前提下,可淋洗出大部分甘油三酯,减少对后续分离检测的干扰。

图1 不同淋洗体积条件的优化
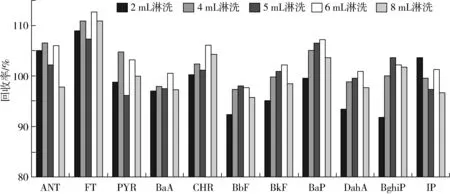
图2 不同淋洗体积时11种多环芳烃回收率
2.1.2 洗脱剂用量的优化
实验中采用中等极性的二氯甲烷作为洗脱溶剂,对洗脱剂用量进行了优化。本实验共使用15 mL二氯甲烷洗脱样品,分三段收集,第一段:0~7 mL;第二段7~10 mL;第三段10~15 mL,并进行液相色谱检测。结果显示,第三段洗脱液中未检出任何多环芳烃组分,第二段洗脱液中检出少量轻质的PAHs,其他组分在第一段7 mL内被洗脱完全。为保证11种多环芳烃能完全被洗脱,本实验选取10 mL做为最佳洗脱体积。
2.1.3 色谱条件的优化
实验中优化了流动相梯度,通过在合适时间改变流动相的比例达到对11种多环芳烃的良好分离(表1),优化确定了各目标物的最佳发射波长和激发波长(表2),11种PAHs的色谱图见图3。
2.2 方法学验证
2.2.1 线性范围、检出限及定量下限
11种多环芳烃的线性范围、检出限及定量限等见表3,配制11种多环芳烃系列标准溶液进行分析。结果表明,在各自线性范围内,相关系数(R2)均大于
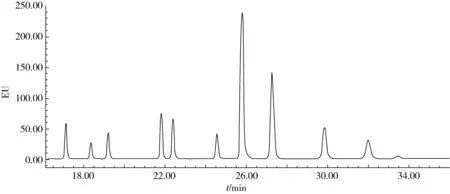
注:从左到右顺序为:蒽、荧蒽、芘、苯并(a)蒽、、苯并(b)荧蒽、苯并(a)芘、二苯并(a,h)蒽、苯并(k)荧蒽、苯并(g,h, i)苝和茚并(1,2,3-c,d)芘。图3 11种PAHs的色谱图

多环芳烃线性范围/ng/mL线性关系相关系数/R2检出限/μg/kg定量限/μg/kg蒽(ANT)0.5~200y= 284 739x+200 8320.999 70.100.33荧蒽(FT)0.5~200y=120 984x+58 9100.999 90.100.33芘(PYR)0.5~200y=253 517x+151 7330.999 70.100.33苯并(a)蒽(BaA)0.5~200y=363 690x+149 3180.999 80.060.20(CHR)0.5~200y=340 938x+171 1160.999 80.050.17苯并(b)荧蒽(BbF)0.5~200y=216 334x+97 0920.999 70.100.33苯并(k)荧蒽(BkF)0.5~200y=1428 669x+487 6410.999 90.030.10苯并(a)芘(BaP)0.5~200y=787 056x-227 8070.999 90.060.20二苯并(a,h)蒽(DahA)1.5~200y= 440 383x+119 5840.999 90.401.30苯并(g,h,i)苝(BghiP)1.0~200y=283 648x+63 6200.999 60.250.83茚并(1,2,3-c,d)芘(IP)5.0~200y=41 398x+13 3920.999 71.505.00
0.999 6,具有非常好的线性关系。对混合标准溶液进行逐级稀释,以3倍性噪比(S/N)计算检出限(LOD),10倍信噪比计算定量下限(LOQ)。可以看出,苯并芘的LOQ明显低于我国植物油卫生标准中对苯并芘10 μg/kg的限量要求,同时也低于欧盟2 μg/kg的限量要求,其他未有限量规定的多环芳烃的LOQ也均小于5 μg/kg,说明本方法灵敏度高,完全满足日常检测的需要。
2.2.2 准确度与精密度
取橄榄油和大豆油空白植物油样品,分别添加高、中、低共4个浓度水平的混合标准溶液,按“2.2样品处理”方法进行处理,每个加标水平进行3次重复实验,添加浓度、回收率及相对标准偏差(RSD)见表4和表5。11种多环芳烃的回收率均在63.5%~118.6%之间,相对标准偏差为1.2%~10.2%。从表4和表5还可以看出,不同品种的植物油,部分多环芳烃的回收率有差距,说明不同品种油脂组分的差别,对淋洗和洗脱效果有影响。以20 μg/kg的大豆油空白加标样品比对本方法和国家标准GB 5009.265—2016《食品安全国家标准 食品中多环芳烃的测定》[3]。本方法测定11种多环芳烃含量在15.3~19.8 μg/kg,加标回收率为83.6%~100.9%,相对标准偏差为0.4%~4.9%;而国标方法检测结果在12.3~18.9 μg/kg,加标回收率为61.7%~94.7%,相对标准偏差为1.2%~15.7%。两种方法的加标回收率、精密度和准确度上均满足GB/T 27404—2008《实验室质量控制规范 食品理化检测》中对检测方法的技术要求,但由于本方法前处理步骤更少,方法快速和简单,因此在方法准确度和精密度上要优于现有国标方法。
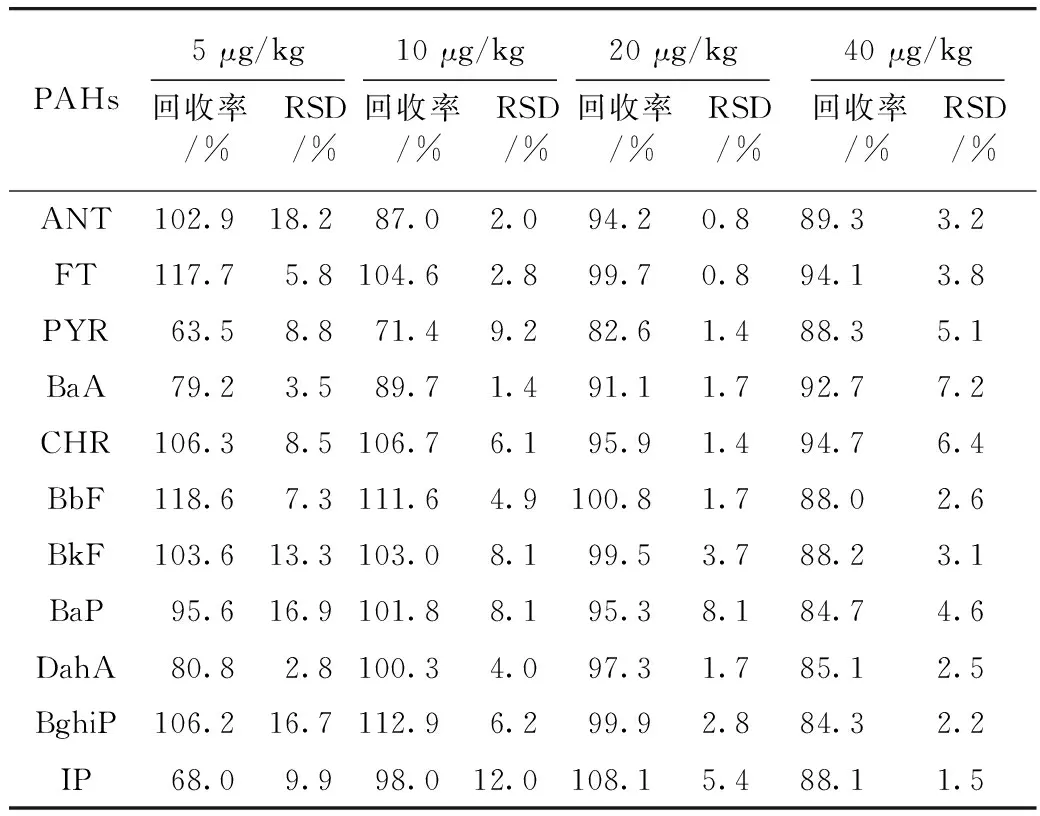
表4 橄榄油11种多环芳烃的加标回收率以及相对标准偏差(n=3)
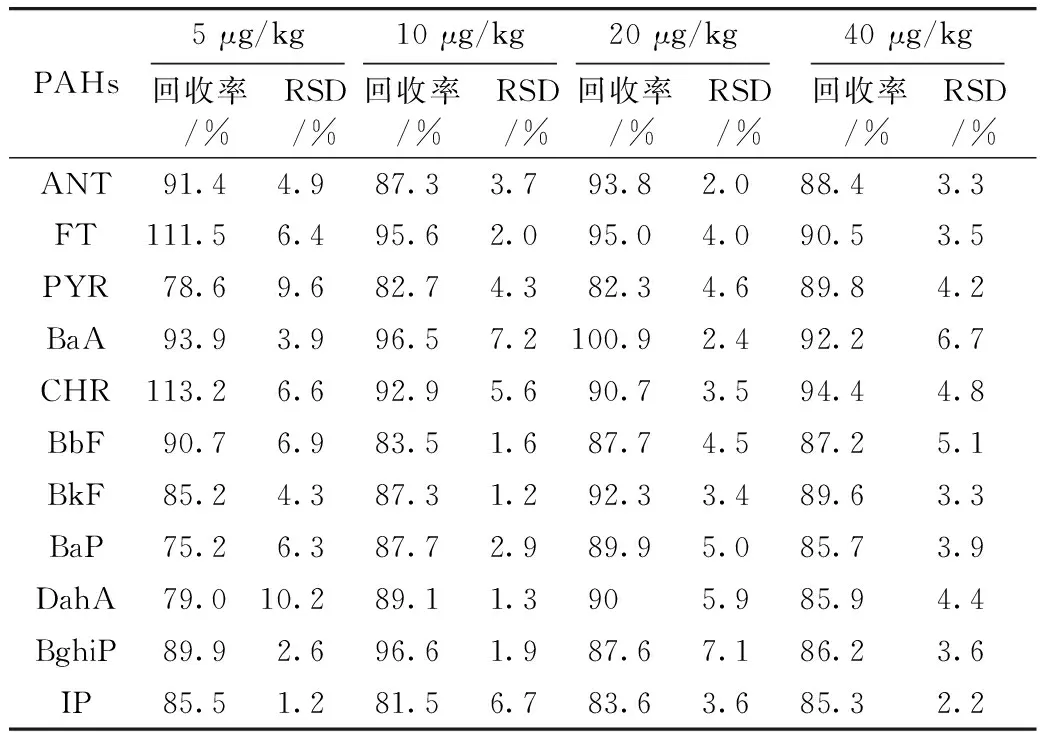
表5 大豆油11种多环芳烃的加标回收率以及相对标准偏差(n=3)
2.3 实际样品的测定
应用本方法对本地超市购买的12个植物油样品进行测定,结果如表6所示,12个植物油中有5种检出苯并(a)芘,苯并(a)芘检出率在42%,所有检出样品的苯并(a)芘含量均符合我国限量要求,但按照欧盟对植物油中苯并(a)芘的限量要求,则有两个样品的结果超标,超标率为16.7%。12个植物油中PAH4均有检出,且PAH4总量超标率高达75%。从结果上看,植物油中含致癌物和2B类致癌物的PAH4污染较为严重,普遍存在,急需加强日常监测。目前我国只对植物油中苯并(a)芘做出限量要求,应加紧制定对植物油中PAH4的限量标准,最大限度保护人民身体健康。

表6 实际植物油样品的11种多环芳烃检测结果/μg/kg
注:ND表示未检出。
3 结论
本研究基于分子印迹固相萃取技术建立了植物油中11种多环芳烃的检测方法,通过采用分子印迹固相萃取,可有效提高多环芳烃在植物油中的提取率,和标准方法相比,大大减少前处理时间和溶剂成本,同时避免复杂前处理过程导致的目标物损失。11种多环芳烃的回收率均在63.5%~118.6%之间,相对标准偏差为1.4%~18.2%。本方法的定量下限符合国内外对多环芳烃的限量要求,可满足大量样品快速、准确定量分析的需要,可用于日常植物油质量安全的检测和监测工作,进一步保证我国植物油质量安全。
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
科学导报(2022年28期)2022-05-24
云南画报(2021年10期)2021-11-24
科学技术创新(2021年19期)2021-07-16
化工管理(2021年7期)2021-05-13
环境保护与循环经济(2020年4期)2020-06-08
中国特种设备安全(2019年1期)2019-03-13
小学生优秀作文(高年级)(2018年4期)2018-09-11
科技创新与应用(2017年1期)2017-05-11
科技与创新(2016年10期)2016-05-28
