
食品级重组产朊假丝酵母菌的构建及其遗传稳定性测定
2019-01-08 05:50其布日萨初拉苏少锋王蕴华刘红葵吴青海
畜牧与饲料科学 2018年12期
其布日,萨初拉,苏少锋,高 娃,王蕴华,刘红葵,吴青海,呼 和
(内蒙古自治区农牧业科学院生物技术研究中心,内蒙古 呼和浩特 010031)
目前绝大多数的酵母表达载体都将大肠杆菌来源的抗性标记和大肠杆菌复制区整合到酵母基因组上,这对于食品安全而言是不利的[1-2]。食品级基因工程表达系统是指其宿主及表达载体为食品工业领域认可的、安全完整的表达系[3]。构建食品级表达系统时用到的宿主菌必须是食品级微生物GRAS,试验材料、抗性标记等都应对环境及人体无毒害作用[4]。食品级工程菌株不能含有非食品级菌种来源的功能性DNA片段[5]。构建食品级表达系统一般有3种方法。第1种方法是通过共转化构建食品级表达载体,即用2种载体:一种是能在宿主菌中复制的载体,另一种是带有抗性标记却不含复制区的伴侣载体,将这2种载体共同转化至宿主菌后,通过表型选择和传代培养获得不含抗性标记的转化子。第2种方法是采用非抗生素抗性选择标记来构建食品级表达载体[6-8]。第3种方法是通过同源重组来构建食品级表达系统,构建载体时会有一些异源微生物的序列,可通过基因工程手段,例如重叠延伸PCR方法删除异源序列,然后将食品级表达系统整合到宿主菌株染色体上,构建食品级载体[9-10]。
重叠延伸PCR方法 (gene splicing by overlap extension PCR,SOE-PCR),即利用具有互补末端的引物,使PCR产物出现重叠链,通过重叠链延伸使不同来源的2条链完成拼接[11]。该方法不用酶切连接处理,引物只要与模板结合,使2段基因出现1个重叠区域,用重叠区域的长引物序列形成重叠链,使基因拼接或重组。SOE-PCR技术在基因点突变、基因敲除、大片段的插入及删除[12]、基因融合等方面有广泛的应用[13-15]。该研究利用SOE-PCR技术将实验室构建的醇溶蛋白基因(zein)酵母表达载体PGZM18中的包括抗药性标记Amp在内的细菌质粒序列DNA片段删除后,重新拼接构建食品级酵母载体。在SOE-PCR中,2个重叠的DNA片段是分别从2个独立的PCR扩增反应得到的,混合2次PCR产物,由于2个突变端引物有重叠区,重叠片段之间退火、延伸成异源双链,加入外侧引物进行第2次PCR,即可得到目标重组体。设计引物时需考虑重叠部分的长度以及几组引物的Tm值相近度,以便第2步混合模板PCR基因融合扩增顺利进行[16-17]。
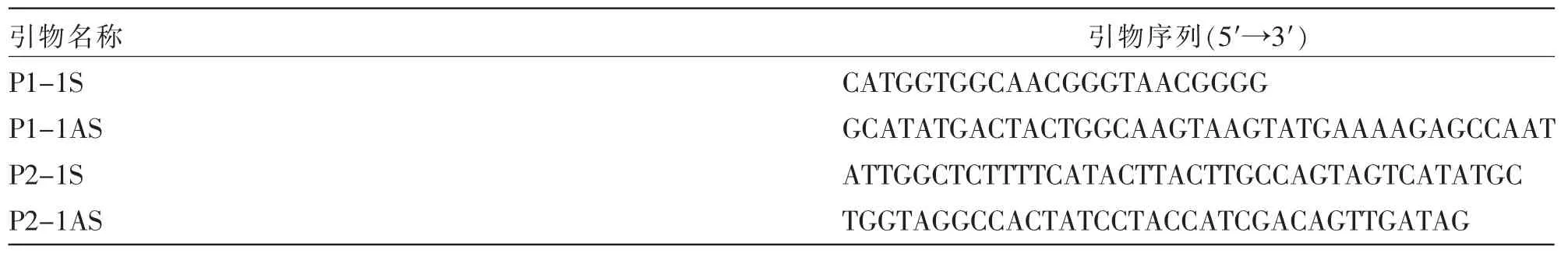
表1 SOE-PCR引物序列
载体中原核基因序列删除后,将真核基因转化至被FDA认定为食品级微生物的产朊假丝酵母中构建食品级工程菌株。由于外源基因转化到宿主中经过多次传代培养易出现外源基因丢失现象,因此,该研究进一步对重组酵母菌株进行了外源基因遗传稳定性的测定。通过开展食品级重组产朊假丝酵母菌的构建及其遗传稳定性的评价,以期为饲料工业中食品级酵母工程菌株的构建以及利用提供科学依据。
1 材料与方法
1.1 试验材料
1.1.1 质粒载体:PGZM18原核载体由笔者所在的实验室构建保存;产朊假丝酵母菌株购自中国工业微生物菌种保藏管理中心,编号为CICC 1769。
1.1.2 主要试剂:三羟甲基氨基甲烷(Tris-碱)、甘氨酸、胰化蛋白胨、酵母提取物均购自生工生物工程(上海)股份有限公司;试验中用到的内切酶、DNA Marker购宝生物工程(大连)有限公司;DNA回收试剂盒 (E.Z.N.A.TMGel Extraction Kit)购自OMEGA公司;引物由南京金斯瑞生物科技有限公司合成。
1.2 试验方法
1.2.1 引物的设计与合成:分析载体质粒序列真核及原核基因部分,以PGZM18原核载体质粒(见图1)为模板,根据参考文献[17]进行引物设计,相关引物序列见表1。
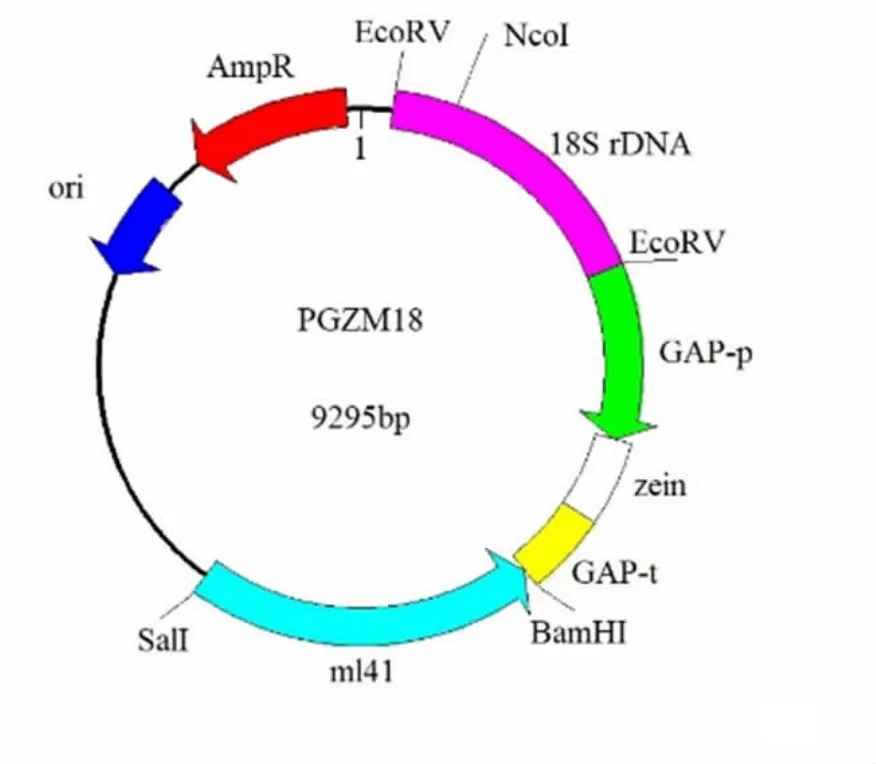
图1 pBR322-GAP-zein-mL41-18S rDNA(PGZM18)载体质粒图谱
1.2.2 扩增P1-1和P2-1基因片段:以PGZM18载体质粒为模板进行引物设计,分别以P1-1S、P1-1AS和 P2-1S、P2-1AS为引物,进行 A、B 2组PCR试验。A组PCR反应条件:94℃预变性1 min;98℃变性 10 s,60℃退火 15 s,68℃延伸 6 min,共30个循环;72℃延伸10 min;4℃保存。B组PCR反应条件:94℃预变性 1 min;98℃变性10 s,60℃退火 15 s,68℃延伸 1 min, 共 30个循环;72℃延伸10 min;4℃保存。A、B 2组各取5 μL PCR产物,用1%琼脂糖凝胶(含1 μL/mL核酸染料)电泳检测,利用紫外凝胶成像系统观察结果,并用DNA回收试剂盒回收目的产物,A、B 2组的PCR产物片段大小分别约为5 085 bp和350 bp。
1.2.3 P1-1和P2-1 2个基因片段的拼接:将P1-1和P2-1 PCR回收产物混合为模板,以P1-1S、P2-1AS作为上、下游引物进行PCR扩增,反应条件为:94℃预变性1 min;98℃变性10 s,60℃退火15 s,68℃延伸6 min,共30个循环;72℃延伸10 min;4℃保存。将混合PCR回收产物命名为Mix1(见图2),并将PCR产物送测序。
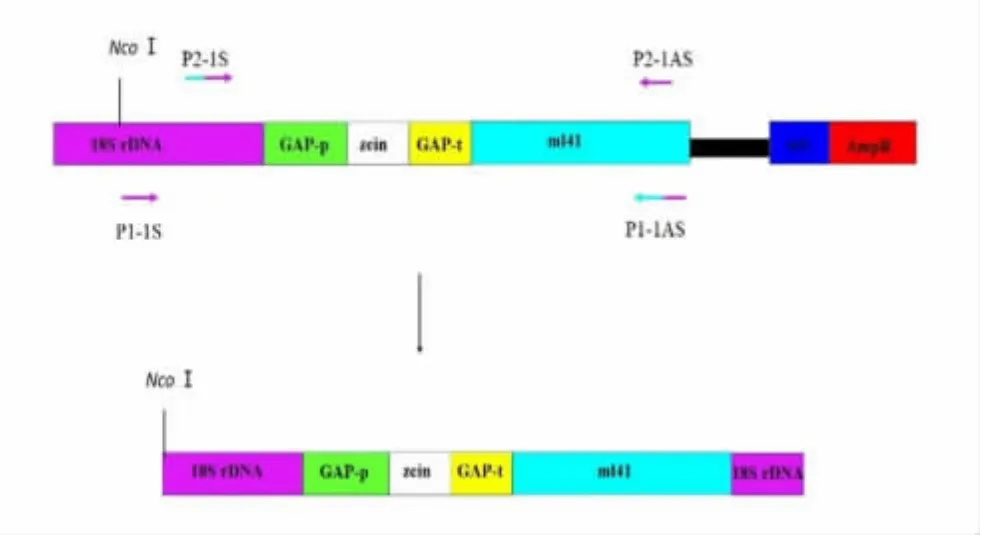
图2 SOE-PCR引物设计以及删除原核序列后的GAP-zein-mL41-18S rDNA(GZM18)结构示意图
1.2.4 删除原核DNA序列后转化至酵母菌株
1.2.4.1 酵母感受态细胞的制备:将保存于-80℃的产朊假丝酵母进行复苏,在YPD固体培养基中30℃培养2~3 d。选择1个单克隆接种于3 mL YPD培养基中,于30℃条件下50 r/min振荡培养8 h。从上一步取5 μL菌液加入50 mL YPD培养基中进行培养,直到菌液OD值达到0.15~0.30(培养16~20 h)。将菌液倒入50 mL离心管中进行离心,弃掉上清液,用新的100 mL YPD培养基重悬沉淀物,然后再进行摇菌培养,直到菌液OD值达到 0.4~0.5(培养 3~5 h)。 将 100 mL 菌液分装到 2个50 mL离心管中离心,弃掉上清液,用30 mL灭菌水重悬,室温离心5 min,弃上清液,加入1.5 mL 1.1×TE/LiAc重悬细胞。将重悬液转到1.5 mL离心管中高速离心15 s,弃上清液,加入600 μL 1.1×TE/LiAc重悬细胞,然后进行PCR产物转化。
1.2.4.2 酵母转化试验:在反应管中加入PCR产物 Mix1 (100 ng/μL)、Yeastmaker Carrier DNA(10 μg/μL)5 μL、酵母感受态细胞 50 μL,轻轻混匀,然后加入500 μL PEG/LiAC轻轻混匀,30℃培养30 min,每隔10 min轻轻混匀1次。加入20 μL DMSO混匀,42℃水浴15 min,每隔5 min轻轻混匀1次,然后高速离心30 s。加入1 mL YPD Plus Medium,30℃培养 90 min,高速离心 30 s,弃上清液,加入1 mL 0.9%NaCl溶液重悬,然后涂布于YPD/CYH(20 μg/mL)平板上,30 ℃培养 3~5 d。
1.2.4.3 酵母重组子的基因组PCR鉴定:对上一步生长在YPD/CYH(20 μg/mL)平板上的重组酵母菌提取基因组,以酵母基因组为模板,设计3组不同的引物鉴定阳性克隆,分别为 Z1、Z2,3S、3AS,5S、5AS。
①根据外源目的基因序列zein设计的引物:引物Z1、Z2的序列及扩增目的片段大小见表2。PCR反应条件为:94℃预变性2 min;94℃变性30 s,53℃退火 30 s,72℃延伸 1 min, 共 30个循环;72℃延伸10 min;4℃保存。 取5 μL PCR产物用1%琼脂糖凝胶(含1 μL/mL核酸染料)电泳检测,利用紫外凝胶成像系统观察结果。
②根据载体序列GAP-zein-mL41(GZM)设计的引物:引物3S、3AS的序列及扩增目的片段大小见表3。PCR反应条件为:94℃预变性2 min;94℃变性 30 s,58℃退火 30 s,72℃延伸 4 min, 共 30个循环;72℃延伸10 min;4℃保存。取5 μL PCR产物用1%琼脂糖凝胶 (含1 μL/mL核酸染料)电泳检测,利用紫外凝胶成像系统观察结果。
③根据载体序列GAP-zein-mL41-18S rDNA(GZM18)设计的引物:引物5S、5AS的序列及扩增目的片段大小见表4。PCR反应条件为:94℃预变性 2 min;94℃变性 30 s,58℃退火 30 s,72℃延伸5 min,共30个循环;72℃延伸10 min;4℃保存。取5 μL PCR产物用 1%琼脂糖凝胶 (含1 μL/mL核酸染料)电泳检测,利用紫外凝胶成像系统观察结果。
1.2.5 外源基因遗传稳定性测定
1.2.5.1 酵母菌落计数:将酵母重组子接种于5 mL YPD培养基中,于28℃条件下100 r/min振荡培养24 h。取1 mL菌液用生理盐水稀释105倍,分别取100 mL菌液涂布于YPD(不含CYH)和 YPD/CYH (20 μg/mL)2 种固体培养基上,28 ℃培养24 h,第2天(记为Day 1)分别计数2种平板上形成的菌落数,YPD(不含CYH)固体培养基的菌落计数结果以 A1 表示,YPD/CYH(20 μg/mL)固体培养基的菌落计数结果以A2表示。将菌液接种于新鲜YPD培养基中,每隔24 h转接1次,连续转接15次,使总的繁殖时间为360 h。每次转接前测定菌液OD600nm值,测定结果以N表示。每次转接后的初始OD600nm值为0.1左右,以使其一直处于生长状态。转化子经过360 h(记为Day 15)生长后进行菌落计数,用同样的方法稀释菌体细胞,并分别涂布于上述2种固体培养基上,待长出单菌落后,菌落计数结果分别以B151和B152表示。质粒稳定性的计算公式为:A2×B151/A1×B152[18-19]。

表2 zein基因引物信息
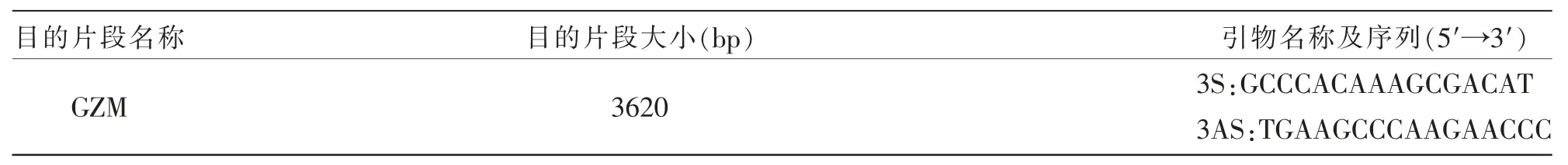
表3 GAP-zein-mL41(GZM)引物信息

表4 GAP-zein-mL41-18S rDNA(GZM18)引物信息

图3 引物P1-1和P2-1 PCR产物电泳检测结果
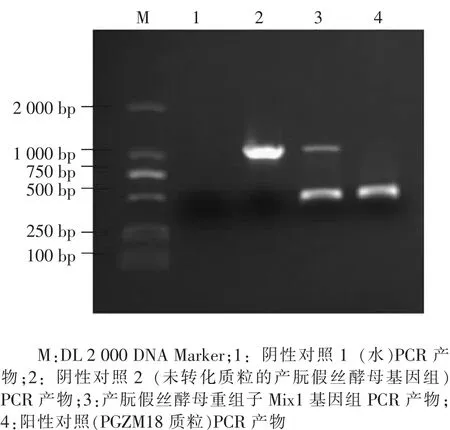
图4 zein基因的PCR鉴定结果
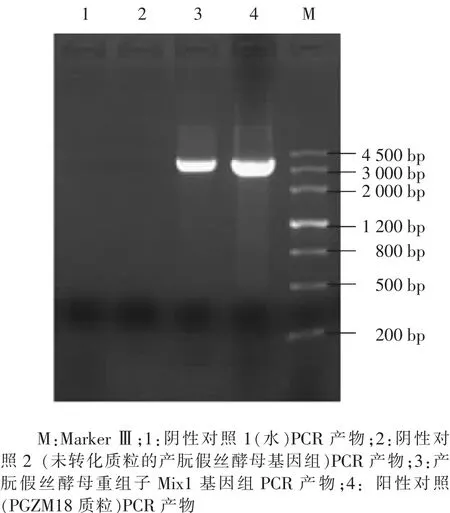
图5 GAP-zein-mL41(GZM)PCR鉴定结果
1.2.5.2 酵母基因组PCR检测:将酵母重组子用上一步的培养方法,分别用YPD(不含CYH)和YPD/CYH(20 μg/mL)2 种固体培养基进行接种传代培养后,选取第1、7、15天的酵母菌培养物提取酵母基因组,利用PCR检测目的基因,测定外源基因遗传稳定性[20]。
2 结果与分析
2.1 P1-1和P2-1 2个基因片段扩增PCR产物鉴定结果
分别利用 P1-1S、P1-1AS 和 P2-1S、P2-1AS引物进行A、B 2组PCR试验。由图3可知,获得的P1-1和P2-1产物片段大小分别为5 085 bp和350 bp左右,与预期扩增大小相符。
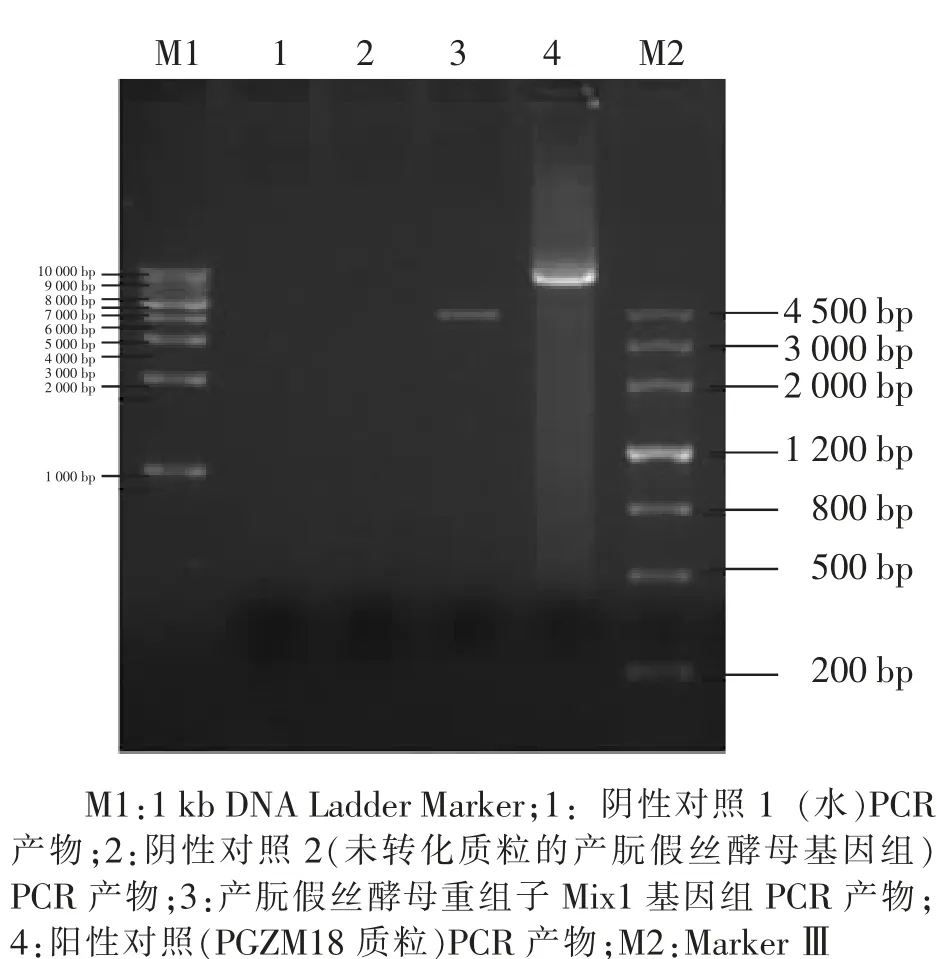
图6 GAP-zein-mL41-18S rDNA(GZM18)PCR 电泳图
2.2 P1-1和P2-1拼接产物的鉴定结果
将P1-1和P2-1的PCR回收产物混合为模板,以P1-1S和P2-1AS分别作为上、下游引物进行PCR试验。获得的P1-1和P2-1拼接产物Mix1的片段大小为5 396 bp。对PCR产物进行测序,结果表明,Mix1的序列与预测的载体真核序列完全一致,表明原核基因序列删除,食品级序列拼接成功。
2.3 酵母重组子鉴定结果
以酵母重组子基因组为模板进行PCR鉴定。zein基因引物Z1、Z2的PCR产物大小为453 bp(见图 4),载体序列 GAP-zein-mL41(GZM)引物3S、3AS 的 PCR 产物大小为 3 620 bp(见图 5),载体序列 GAP-zein-mL41-18S rDNA(GZM18)引物5S、5AS的PCR产物大小为4 446 bp(见图6)。
2.4 外源基因遗传稳定性测定结果
2.4.1 菌落计数方法检测遗传稳定性:外源基因在宿主产朊假丝酵母基因组上的遗传稳定性检测结果见图7,不同培养时间及不同培养基中的酵母菌落计数结果见表5。由表5可知,整合型重组质粒在酵母的基因组中有良好的稳定性,培养100代左右,外源基因在重组酵母中仍能够稳定存在。根据表5中列出的相关数据,利用上述质粒稳定性计算公式,得到质粒稳定性结果为0.93。
2.4.2 PCR检测目的基因鉴定遗传稳定性:提取重组酵母基因组DNA,利用zein基因引物进行PCR扩增。由图8可知,目的基因稳定存在,在酵母菌传代100代时仍含有目的条带,表明外源基因没有丢失,在传代培养过程中保持了较高的遗传稳定性。
3 讨论与结论
该研究利用SOE-PCR技术,删除原有载体PGZM18中包含抗药性标记Amp在内的细菌质粒序列DNA片段及原核复制区序列;将真核基因序列通过同源重组技术转化至产朊假丝酵母染色体中;重组酵母菌株经过多次传代培养,通过菌落计数和外源基因PCR方法鉴定整合到染色体上的外源基因具有较好的遗传稳定性,从而获得遗传稳定的食品级酵母工程菌株。食品级基因工程菌在工业生产的应用日益增多,而在食品、医药、饲料等领域的应用则需要更安全的食品级工程菌株。然而,构建的食品级工程菌株能否最终应用于工业领域,使重组酵母菌株进行大规模的培养以及外源基因的大量表达等还需更多的深入研究。
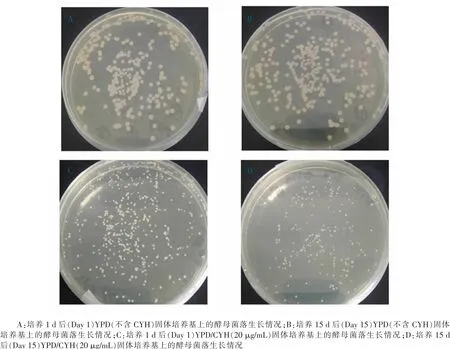
图7 遗传稳定性试验中不同培养时间及不同培养基中的酵母菌落生长情况
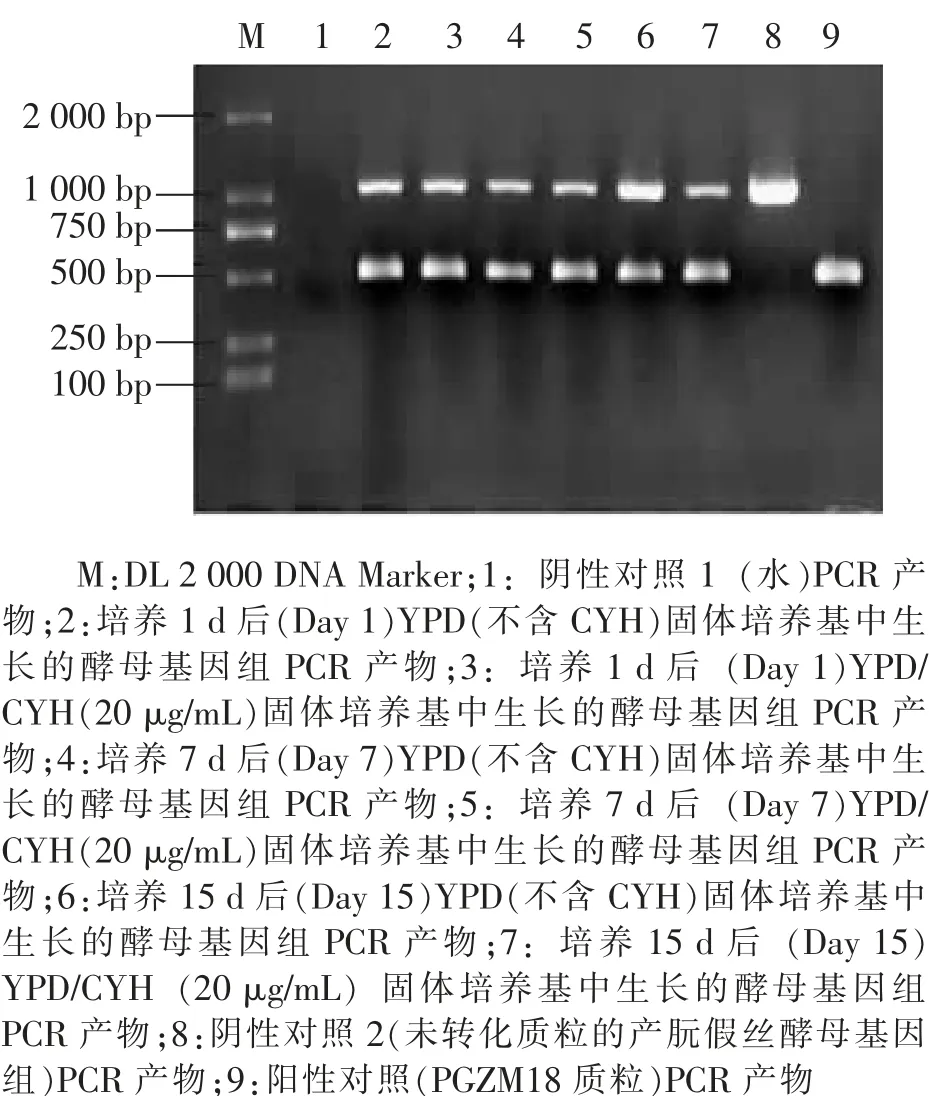
图8 外源基因zein遗传稳定性PCR检测结果

表5 不同培养时间及不同培养基中的酵母菌落计数结果
猜你喜欢
舰船科学技术(2022年11期)2022-07-15
昆明医科大学学报(2022年1期)2022-02-28
文萃报·周五版(2021年37期)2021-10-09
农业资源与环境学报(2021年5期)2021-10-06
军事文摘·科学少年(2021年1期)2021-02-04
当代水产(2019年3期)2019-05-14
天然产物研究与开发(2018年9期)2018-10-08
中国化妆品(2018年5期)2018-06-28
现代园艺(2018年3期)2018-02-10
上海农业学报(2017年3期)2017-04-10
